Team:Heidelberg/Parts
Parts
Discover our workpieces and standards

Our Best Basic Part: Cytochrome C
BBa_K2398000
We present as best basic part the codon-optimized version of the cytochrome c protein derived from Rhodotermus marinus that is able to convert silicon educts to organosilicon products.
As a next step, this part can be implemented in the directed evolution approach of phage-assisted continuous evolution (PACE) or in the phage-related discontinuous evolution (PREDCEL) approach to improve organosilicon synthesis by cytochrome engineering. For the characterization of this part, we conducted experiments for the production of organosilicons and therefore used a previously described triple mutant, already engineered by [Arnold et al., 2016]. According to the protocol, we obtained the matured cytochrome c and performed conversion experiments analyzed via GC-MS method.
The following figures show the results obtained with the previously engineered enzyme. First, we tested the enzyme on the conversion of the compound (1) with (3) to the final product of (5) (Fig. 1). The gas chromatogram shows one product peak which emerges at 9.2 min retention time. Analysis of the gas chromatogram indicates a conversion rate of >99% on the basis of the non-existence of the respective educt peak. As we performed GC-MS analysis, mass spectrometry data were also available which can be seen in Fig. 3 for the product peak that emerges at 9.2 minutes. It shows the fission of this product and can clearly be identified as our desired product (5). Furthermore, we performed experiments on the compound (1) which differs from compound (2) in its amino functional group. The product is eluted after 11.7 minutes retention time (Fig. 4) and also confirmed as the right product via mass spectrometry in Fig. 5. Worth mentioning, there is still educt (1) present which emerges at 6.9 minutes retention time, indicating incomplete conversion. Calculations showed a conversion rate of 47.5% for the product (4).As these results only show activity for a previously engineered cytochrome c, we are convinced that our part is still functional despite not as effective as the mutant. This part as our best basic part is intended as a platform for future teams to evolve their own cytochrome c for their specific target molecule.

Figure 1:
Compounds used in the project of organosilicons and its respective products.

Figure 2:
Gas chromatogram for the reaction of educt (1) and (3) to the product (5). 11.7 minutes retention time indicates product formation. Unconverted educts converge 6.9 and 7.2 (7.4) minutes.

Figure 3:
Mass chromatogram shows the breakdown of the product (5). The product itself corresponds to a mass of 251 daltons.

Figure 4:
Gas chromatogram for the reaction of educt (2) and (3) to the product (4). 9.2 minutes retention time indicates product formation.

Figure 5:
Mass chromatogram shows the breakdown of the product (4). The product itself corresponds to a mass of 236 daltons.
Our Best Composite Part
BBa_K2398019: Theophylline Riboswitch - GeneIII for the Evolution of Enzymes
One of our main goals was to facilitate for the in vivo directed evolution of small molecule binding enzymes. The concept behind this idea is the use of riboswitches for the activation of geneIII. To prove our idea, we made use of a theophylline binding riboswitch in relation with the Enzyme CYP1A2, which catalyzes the reaction of caffeine to theophylline. As we could show that the principle of enzyme evolution with the help of riboswitches works, our best composite part is the theophylline riboswitch, followed by geneIII and flanked ba homology regions, which make it compatible with our cloning standard.Background

Figure 1: Selection process for improved CYP1A2 variants in directed evolution experiments
The evolutionary circle starts by M13 phages injecting their genome (SP) into bacterial cells, already containing two additional plasmids, AP and MP. The SP encodes CYP1A2 among genes (except geneIII) that are crucial for phage propagation. If through MP activation mutations in the CYP1A2 gene lead to improved CYP1A2 variants the intracellular level of theophylline increases. Theophylline molecules activate the theophylline riboswitch on the AP and thereby enhance geneIII expression. The assembled phages containing the improved CYP1A2 variant can leave the cell and propagate by infecting new cells.
Our evolution circuit for cytochrome engineering works as follows: Bacteriophages infect bacterial cells by introducing their genome. The genome encodes a Selection Plasmid (SP) that contains the human CYP1A2 variant and all necessary components for virus propagation except geneIII. The Accessory Plasmid (AP) codes for geneIII driven by a Psp-tet promoter and contains a riboswitch, located between the promoter and geneIII, which regulates the expression rate of geneIII. The riboswitch on the AP is only active if theophylline reaches a certain concentration within the bacterium. If CYP1A2 is active, caffeine is converted to theophylline. Gradually the theophylline concentration increases and acts upon the riboswitch, which enables expression of geneIII. Additionally, the AP encodes for the chaperone HDJ-1, which is essential to receive the functional CYP1A2 enzyme (Fig. 1).
In a first step, we wanted to validate our AP. Therefore, we added theophylline with a concentration of 100 µM to our inoculated culture and performed two rounds of PREDCEL. Afterwards, we determined the phage titers by plaque assays. Our theophylline treated culture displayed approximately two times higher phage titers than the non-treated control culture.
Using the same experimental conditions, but replacing the theophylline treatment by a 300 µM caffeine treatment, we verified the functionality of CYP1A2 and thus of our SP. If caffeine is added to the culture, CYP1A2 catalyzes the reaction from caffeine to theophylline. The resulting increase of the theophylline concentration further activates the riboswitch on the AP and phage propagation is stimulated (Fig. 2).
For the evolution of proteins via PREDCEL the addition of a Mutagenesis Plasmid (MP) is essential. For our cytochrome engineering approach we have chosen MP4, which induces a medium mutation rate badran2015development . After six iterations of our optimized PREDCEL workflow, we performed plaque assays and sequenced single plaques. The sequenced plaques showed five recurrent mutations demonstrating that we are able to induce mutations with our experimental setup and that we are able to evolve enzymes (Fig. 7).

Figure 2:
Plaque assays performed after two passages of CYP1A2 PREDCEL
with either adding 100 µM theophylline or 300 µM demonstrating the
functionality of the Accessory Plasmid and the Selection Plasmid,
respectively. Adding theophylline increases the geneIII expression
2-fold. Adding caffeine enhances the conversion of caffeine to
theophylline and thus increases the geneIII expression as well.

Figure 3:
Sequencing results of eight plaques after 6 iterations of
the PREDCEL workflow with MP4 illustrating recurring
mutations of the CYP1A2 gene. Recurrent mutations with amino acid
exchange are indicated in red, without amino acid exchange in orange.
Single mutations with amino acid changes are shown in yellow, and
without amino acid changes in blue.

Figure 4:
Cell lysates were used for high-performance liquid chromatography to
distinguish theophylline (left peak) from caffeine (right peak). This
assay allows a quantification of the educt and the product of CYP1A2
and thus a conclusion about the conversion efficiency can be made.
Improved Part
Engineered highly active Beta-Lactamase mutant E102F
This part is an improvement of BBa_K1189009: Beta-Lactamase with His-Tag.The aim of our project was to generate protein functionality, in vivo through PACE and PREDCEL and in in vitro with help of our software suite AiGEM. To prove that our circuit works, we mutated ß-lactamases based on the scores of our software. Many different mutants were cloned and screened for their minimal inhibitory concentration (MIC). Visit our software validation site here. Typical methods to determine MICs are broth or agar dilutions. The principle is as simple as effective. The microorganism is cultured in liquid medium or on plates with different concentrations of the inhibiting agentIn our case, we want to evaluate differences in the activity of a well characterized protein. Consequently, such an dilution method is ideal. It is easy to setup, very robust and gives exactly the information that is needed for the characterization.
To start the assay, colonies were picked into LB medium with 100 µg/ml chloramphenicol, but without carbenicillin and were grown to the stationary phase for 20 h. Then, deep well plates with 500 µl LB broth per well were prepared. The medium was supplemented with 100 µg/ml chloramphenicol and for each mutant, eight wells with different carbenicillin concentrations, ranging from 10-1280 µg/ml were prepared. Each well was inoculated with 2 µl of the proculture. The new cultures were incubated another 8 h. Finally, the OD600 of each well was measured. By this measure we were able to assert the growth ability of each variant. A first screening revealed, that beside catalytically inactive variants, we also had many candidates that even could survive the highest carbenicillin concentrations (1280 µg/ml)(Fig.: 1). As a result, we decided to perform a second test for these candidates with higher antibiotic concentrations. The three highest carbenicillin concentrations of the first data set were included and the range was extended to a maximum concentration of 19.2 mg/ml. While several candidates turned out to have a weaker activity than the wildtype protein, five of them showed improved properties. The two best candidates could even grow at 19.2 mg/ml (Fig.: 2). The variants were contained one point mutation each, E102F and M67G. These most benefitial mutations, appear in many of our better candidates as well, which underlines there functionality. The outstanding candidate was the E102F mutant, which could even grow at 19.2 mg/ml without affection by the antibiotic. This results in a improvement of at least 100 % in comparison to the wildtype ß-lactamase. This remarkable gain of activity proves, that our software is not only able to recognize protein classes, but also to generate new function.
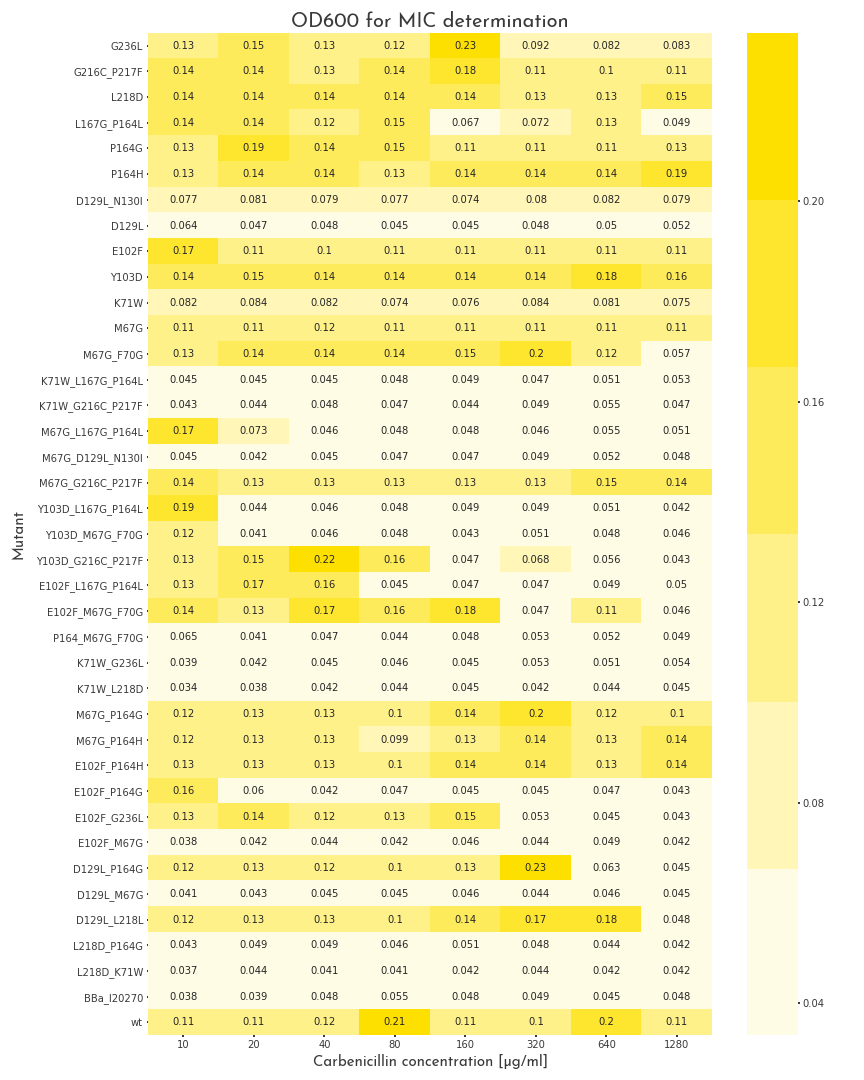
Figure 1: Gowth Behaviour of \(\beta\)-Lactamase Mutants under Different Carbenicillin Concentrations
The plot shows the OD600 of the different \(\beta\)-lactamase mutants at different carbenicillin concentrations. The pool of mutants is very heterogenous. Some proteins are not active at all, some show different grades of activity and 11 mutants, as well as the wildtype enzyme can grow in all conditions.

Figure 2: Gowth Behaviour of \(\beta\)-Lactamase Mutants under Elevated Carbenicillin Concentrations
The plot depicts the \(OD_{600}\) of the \(\beta\)-lactamase mutants that could survive at 1280 \(\frac{µg}{ml}\) in the first assay, under more stringent conditions. The majority of these \(\beta\)-lactamases have a slightly weaker activity than the wildtype, but we could determine five variants with a higher enzymatic activity.
Parts Collection
We present the first self-contained in vivo evolution toolbox comprising 26 standardized BioBricks. The collection includes several key aspects of our project:The crucial step of to the cloning of PACE circuits is the generation of the accessory plasmid. These plasmids allow geneIII expression dependent on the evolving protein. The link between the fitness of the protein of interest and the expression of geneIII determines the effectiveness of the directed evolution and the presence of ProteinIII is essential for the production of new phage particles.
Therefore, we provide an accessory plasmid construction kit for phage library selection and propagation in E. coli based on conditional geneIII complementation. The geneIII-driving promotor, RBS and plasmid ori can thereby be fully customized.
If more than one variant of a circuit should be tested at a time, it is necessary to modify the activation region for geneIII and the additional gene that can be located on the AP with a minimum of effort. This is the another reason for a efficient cloning strategy.
To make AP cloning as simple as possible, we defined a new cloning standard that is specifically suited for the assembly of APs, we wrote a BBF RFC that describes our concept in detail (Fig. 1).

Figure 1: In our cloning standard, compatible building blocks are defined by specific functionalities. They are flanked by defined homology regions,
indicated by numbers, which are necessary for the assembly of the APs with the Gibson method. This
results in a highly customizable plasmid, composed of the desired origin of replication, an antibiotic
resistance (4-5), a bicistronic operon with geneIII (2-3)and the desired reporter (3-4), which can be
activated by any promoter (1-2)and a second expression cassette for additional genes that are necessary
for the respective circuit (1-5).
In Addition, an optogenetic selection stringency modulator comprising the light-dependent EL222 transcription factor and an engineered, hybrid Psp-EL222 promoter.
Furthermore, we provide parts for production of geneIII-deficient M13 phages carrying a custom gene-of-interest to be evolved.
On order to validate our system, we constructed the plasmids for our CRISPR/Cas9 subproject according to the RFC cloning standard. We could constuct several plasmids with help of this standard.
At the heart of our toolbox is an innovative workflow for enzyme engineering, paving the way towards bio-production of novel compounds. It comprises promiscuous enzyme candidates (CYP1A2 and Cytochrome c) and selection-mediating accessory parts such as our riboswitch for detection of an organosilicon (BBa_K2398555). Our simple PREDCEL protocol and accompanying online toolbox guide bring in vivo evolution to future iGEM teams and the broader synbio community.
Finally we created a set of parts, which were used in our different subprojects. These parts were transferred into pSB1C3 in a way that they are still compatible with our standard and submitted them to the registry:
Loading...
Parts List
Loading...