Team:UMaryland
An Apeeling Solution to Panama Disease
Projects
Saving the Cavendish banana
Outreach
Increasing access to synthetic biology for high schools
PartsModelingNotebook
About Us
Learn more about UMaryland iGEM
← Projects
From saving the Cavendish to expanding synthetic biology
An Apeeling Solution
to save the Cavendish
Cas9 Mutant Screening
Screening for mutants using CRISPR/Cas9
Lab-in-a-box
Low cost DIY lab equipment
Metal Detection
Teaching synthetic biology using real world examples
← Outreach
Reaching out to the community about synthetic biology
Knowing Panama Disease
Feedback, concerns, and feasiblity
Teaching the Next Generation
of synthetic biologists
Talking with the Community
about synthetic biology
← About Us
UMaryland iGEM - Since 2014
Students
The next generation of scientists
Advisors
Guiding our efforts
Funding
Providing support for the team
Acknowl-edgements
Those who helped us get here
← Application
Contributing to the scientific community
Parts
Contributions to the Registry
Modeling
Applying engineering principles
Collabor-ations
Working together with other synthetic biologists
Notebook
Follow our progress throughout our experience
The banana is the most popular fruit in the world, with a whopping 160 million tons produced annually. In America, we think of the banana as a fruit, something we love but could live without. However, the same cannot be said for over 400 million people in developing countries. These individuals rely on bananas as their biggest, and sometimes, sole source of calories and nutrition. Terrifyingly enough, 7% of the world’s population could lose its sole source of food as the banana crop is being ravaged by a devastating pandemic: Panama disease.

Panama disease is caused by the fungus Fusarium oxysporum f. sp. Cubense race 4, which is deadly to the Cavendish banana. This fungus is able to first infect the roots of the plant, secreting different hydrolytic enzymes, and subsequently infect the xylem, blocking water and nutrient flow, leading to the death of the plant. Symptoms, however, are typically not visible until four months after infection, providing ample time for the fungus to spread without notice. Additionally, the fungus remains in the soil in the form of spores for up to a decade, ready to infect the next set of plants. Therefore, there is a need to develop a method of protecting the Cavendish banana from Fusarium oxysporum and eliminating the fungus from the soil.
Many eukaryotes produce a class of proteins known as thaumatin-like proteins (TLPs) in response to attack and stress. Some of these TLPs are known to have antifungal properties against F. oxysporum. Among these are the TLP from Oryza sativa (rice) and Arachis diogoi (wild peanut plant).
Current methods of fungal inhibition are too detrimental and cannot be used for extended periods of time. However, recent studies have shown that biocontrol methods using disease-suppressive soils have been quite effective at fungal inhibition. Bacillus amyloliquefaciens (BA) NJN-6 is a common Bacillus strain found in the Cavendish banana rhizosphere. BA has been found to produce some antifungal compounds that are fairly effective at suppressing fungal growth. By combining BA with compost to make a fertilizer and applying it to soil already infected by F. oxysporum, the susceptibility of the banana plants to the fungus was greatly reduced. While quite effective, the bacteria alone is unable to kill the fungus.
As a result, we propose to transform BA to produce an antifungal TLP. Then we will again combine this with compost to create a fertilizer that is even more potent. Since TLPs are known to actually be able to kill the fungus in addition to being able to inhibit spore germination, this transformed BA has the potential to be more effective against F. oxysporum.

Bacillus amyloliquefaciens, a bacterium that grows synergistically with the roots of the Cavendish banana plant, senses Fusarium oxysporum, the fungus causing Panama disease. In response, B. amyloliquefaciens produces TLP, an antifungal, to fight off the pathogen.
There is currently a lot of stigma around and distrust of genetically modified foods. As a result, we wanted to focus on creating a genetically modified organism that would serve as a probiotic treatment for banana plants.
There is currently a lot of stigma around and distrust of genetically modified foods. As a result, we wanted to focus on creating a genetically modified organism that would serve as a probiotic treatment for banana plants.
Our initial steps will involve making plasmid designs for E. coli (as E. coli will the the initial chassis used for proof of concept). Our E. coli must be able to sense F. oxysporum. Upon presence, the bacteria should produce the antifungal TLP and there should be a mechanism for the TLP to be able to interact with the fungus. In addition, the E. coli must also be able to survive in the conditions that result from the presence of the fungus. In summary, the basic components of our circuit are a F. oxysporum sensing system, a fungal killing mechanism, and a chassis survival mechanism.

We wanted to find a way to uniquely sense F. oxysporum, such that a response is only triggered in its presence. We found that F. oxysporum produces fusaric acid, a small weakly acidic (pKa 1.1) mycotoxin and phytotoxin that is able to diffuse across the cellular membrane. Fusaric acid contributes to the virulence of the fungus. It causes super polarization, suppressed H+ pumping, and K+ leakage in plant cells. It also reduces oxygen absorption by mitochondria and causes malic acid oxidation. Downstream effects include the creation of reactive oxygen species and changes in membrane polarity and permeability . Fusaric acid is also toxic to several bacterial species, including E. coli, posing a possible issue for testing that we addressed later on.
Different bacterial species have shown unique sensitivities to fusaric acid. Some organisms that are resistant to the compound are able to produce efflux pumps in its presence. These include Pseudomonas putida and Stenotrophomonas maltophilia, which both incorporate an efflux system that is repressed when there is an absence of fusaric acid. Our initial plans were to design a plasmid with TLP regulated by a promoter that was repressed by these fusaric acid-sensitive repressors. However, the operators have not yet been identified, making it difficult to adjust the system to E. coli.
Fusaric acid has also been shown to suppress the production of an antimicrobial compound, 2,4-diacetylphloroglucinol (DAPG), produced in Pseudomonas fluorescens. In this system, fusaric acid is able to induce the phlF repressor that is then able to suppress expression of the adjacent phlA gene. Concentrations of 500uM completely repress transcription, 300 uM produce some inhibition, and 100 uM have no effect. In 2015 Glasgow combined the characterized operator of the phlF sensitive promoter native to P. fluorescens and incorporated it into a strong E. coli Anderson promoter. We will use both the repressor and synthetic promoter in our design to make our bacteria sensitive to fusaric acid. The DNA following this part will have suppressed transcription in the presence of fusaric acid.

The natural system follows the sensing of F. oxysporum with inhibition of the next component, which was opposite of the circuit we wanted. Therefore, we created an intermediate step in the circuit that turned inhibition into activation by having the phlF repressor act on a gene responsible for repressing the killing mechanism in normal conditions.
The killing mechanism required us to produce an antifungal that will target F. oxysporum. For this part, we chose to incorporate rice thaumatin-like protein (TLP) into our design. Plant TLPs are defense pathogenesis related proteins of family 5. Rice TLP has shown antifungal activity both when over-expressed in the rice plant and also when expressed in E. coli. The exact mechanism is still being studied, but TLPs are suspected to disrupt the cell walls of fungi by binding to glucans and acting as glucanases. β-glucan is the most abundant polysaccharide found in fungal cell walls and its degradation destabilizes the fungal cell wall. TLP may also provide resistance to fungal disease as a xylanase inhibitor, inhibiting an enzyme that breaks down a component of plant cell walls. Because literature sources have confirmed the antifungal properties of rice TLP, as well as its ability to be produced by E. coli, we chose to use this protein as our fungal attack piece.
Amounts of 5 ug of rice TLP extracted from recombinant E. coli were enough to inhibit fungal growth of 7 mm mycelial discs and concentrations of 500 ug / mL of TLP showed over eighty percent inhibition of spore germination of F. oxysporum f. sp cubense.
From the components we had gathered through research, we created three initial design ideas.
| Design | Pros | Cons |
| TetR | Simple, easiest to construct | Low expression of TLP (weaker promoter), iffy time scale (would TetR be degraded fast enough) |
| Antisense | Pretty neat idea, more novel, quick RNA degradation rates, smaller plasmid | Need correct ratio - causes need to limit TLP transcript number, slightly harder to make |
| T7 | Optimized for production - T7 very fast, efficient polymerase | Higher metabolic toll, bigger plasmid |
| All | Unsure if E. coli will survive before production of TLPUnsure if TLP will be able to go outside of cellsUnsure if amount TLP produced will show significant changes in fungal growth |
All three of these designs have the same logical format: If there is no fusaric acid in the environment, something will inhibit TLP production. When fusaric acid is present, that inhibition will be taken off and expression will occur.
After some mathematical modelling and assessments, we decided that the antisense system was the best option, and so it was incorporated into our final design.
Additionally, we initially predicted that eventually the overexpression of the TLP will kill the cell causing TLP release to the exterior surroundings. This TLP would be available to interact with and attack the fungi. However, we were advised that this method was not reliable due to the possibility of protein aggregation and so we decided to secrete the protein instead. We found several iGEM teams had worked with protein secretion in E. coli, including Unicamp - EMSE Brazil 2011 and used the information they provided as well as their results to create a secretion part. We found that gram negative bacteria, like E. coli, have a secretion system that involves HlyB, HlyD, and TolC proteins. HlyB is an ATP-binding cassette transporter. HlyD is a membrane fusion protein, and TolC is an outer membrane protein. HlyB and HlyD are not present in all strains, so there was a necessity to express them in our bacteria and incorporate the two proteins into our part. In order for a protein of interest to be secreted through this system, it must be labeled with an HlyA signal sequence, which we decided to add to the C terminus of our TLP.
We integrated all the different parts and components to one design.


We have also considered adding a fusaric acid detoxification component into our system in efforts to promote the survival of the E. coli. It was discovered that Klebsiella oxytoca has a three enzyme detoxification system that was identified and sequenced, fdt-1, fdt-2, and fdt-3. While it may be needed for testing purposes later, as the survival of E. coli in our induced conditions is invaluable, this part will not have an analog in our Bacillus amyloliquefaciens system, as B. amyloliquefaciens has its own mechanisms of protection.
As we hope to apply this basic design to a B. amyloliquefaciens vector, we also designed an analogous system. We found proposed sequences for promoters and ribosome binding sites native to B. amyloliquefaciens to swap out our E. coli sequences. For our sensing mechanism we plan to incorporate the operator phlF interacts with upstream of the antisense component. The same paper provided a proposed signal sequence for export used on the n-terminus of an amylase gene. We propose to put this sequence on the n-terminus of our TLP gene and remove the entire secretory system of our E. coli design. We also found a proposed terminator sequence. We plan to codon optimize the protein coding regions and adjust the antisense accordingly.
Bibliography
- Chunyu Li, Cunwu Zuo, Guiming Deng, Ruibin Kuang, Qiaosong Yang, Chunhua Hu, Ou Sheng, Sheng Zhang, Lijun Ma, Yuerong Wei, Jing Yang, Siwen Liu, Manosh Kumar Biswas, Altus Viljoen, and Ganjun Yi, Contamination of Bananas with Beauvericin and Fusaric Acid Produced by Fusarium oxysporum f. sp. cubense. PLoS ONE 8 (2013), no. 7.
- O’Neill, W. T., Henderson, J., Pattemore, J. A., O’Dwyer, C., Perry, S., Beasley, D. R., and Shivas, R. G., Detection of Fusarium oxysporum f. sp. cubense tropical race 4 strain in northern Queensland. Australasian Plant Disease Notes (2016), 11(1), 33.
- Anne Vézina, Fusarium wilt of banana. Knowledge and Information on Bananas (2017)
- Rouh Mei Hu, Sih Ting Liao, Chiang Ching Huang, Yi Wei Huang, and Tsuey Ching Yang, An Inducible Fusaric Acid Tripartite Efflux Pump Contributes to the Fusaric Acid Resistance in Stenotrophomonas maltophilia. PLoS ONE 7 (2012), no. 12, 1–8.
- Regina Notz, Monika Maurhofer, and Helen Dubach, Fusaric acid-producing strains of Fusarium oxysporum alter 2, 4-diacetylphloroglucinol biosynthetic gene expression in Pseudomonas fluorescens CHA0 in vitro. Applied and Environmental Microbiology 68 (2002), no. 5, 2229–2235.
- Jun Jun Liu, Rona Sturrock, and Abul K.M. Ekramoddoullah, The superfamily of thaumatin-like proteins: Its origin, evolution, and expression towards biological function. Plant Cell Reports 29 (2010), no. 5, 419–436.
- J Jayaraj, R Velazhahan, D Fu, G H Liang, and S Muthukrishnan, Bacterially produced rice thaumatin-like protein shows in vitro antifungal activity, Zeitschrift fur Pflanzenkrankheiten und Pflanzenschutz. Journal of Plant Diseases & Protection 111 (2004), no. 4, 334–344.
- Su, L., Chen, S., Yi, L., Woodard, R. W., Chen, J., & Wu, J., Extracellular overexpression of recombinant Thermobifida fusca cutinase by alpha-hemolysin secretion system in E. coli BL21(DE3). Microbial Cell Factories (2012), 11, 8.
- H. Toyoda, K. Katsuragi, T. Tamai, and S. Ouchi, DNA Sequence of Genes for Detoxification of Fusaric Acid, a Wilt-inducing Agent Produced by Fusarium Species. Journal of Phytopathology 133 (1991), no. 4, 265–277.
- Palva, I., Pettersson, R. F., Kalkkinen, N., Lehtovaara, P., Sarvas, M., Söderlund, H., Kääriäinen, L., Nucleotide sequence of the promoter and NH2-terminal signal peptide region of the alpha-amylase gene from Bacillus amyloliquefaciens. Gene (1981), 15(1), 43–51.
- Lehtovaara, P., Ulmanen, I., & Palva, I., In vivo transcription initiation and termination sites of an alpha-amylase gene from Bacillus amyloliquefaciens cloned in Bacillus subtilis. Gene (1984), 30(1–3), 11–16.
As discussed in the Design Section of the Banana project, we devised three possible designs to transduce the fusaric acid signal. Here’s a quick recap of the designs:
- The TetR Design places TLP expression under a pTet promoter. TetR is expressed under the phlF promoter. In the presence of fusaric acid, TetR does not express, allowing for TLP to express.
- The Antisense Design places TLP expression under a constitutive promoter. An antisense RNA that binds to the TLP transcript is expressed under the phlF promoter. In the presence of fusaric acid, the antisense RNA is not produced, and the TLP transcript is liberated and translated into TLP.
- The T7 Design places TLP expression under a T7 promoter. T7 polymerase is expressed under the pTet promoter. TetR is expressed under the phlF promoter. In the presence of fusaric acid, TetR does not express, allowing T7 polymerase to express, leading to the expression of TLP.
The overall question we’re trying to answer is: which design is best? In order to answer this question, we broke it down into more concrete questions that we could search in the literature.

Following this roadmap, here is what we found:
- How do the promoters stack up in terms of strength?:
- According to the 2015 Glasgow iGEM team, the phlF promoter is slightly stronger than the pTet promoter (Figure 1). According to literature1, the T7 promoter should be about three times as strong as the phlF promoter.
 Glasgow iGEM 2015
Glasgow iGEM 2015
- According to the 2015 Glasgow iGEM team, the phlF promoter is slightly stronger than the pTet promoter (Figure 1). According to literature1, the T7 promoter should be about three times as strong as the phlF promoter.
-
How fast is the signal? This question comes down to the time it takes for each of the signal molecules to degrade. The TetR Design and the T7 Design rely on the degradation of TetR to begin TLP expression. The Antisense Design relies on the degradation of an antisense RNA molecule to begin TLP expression.
- TetR degrades in ~40 minutes when fitted with an LVA tag.2
- Antisense RNA usually degrades in ~8 minutes.3
- How toxic is fusaric acid?
- The numbers varied, but we found that fusaric acid has a strong impact on the cell. Fusaric acid inhibits growth of Bacillus Subtilis by 60% at a concentration of 200 ug/ml.4 Future work of our group includes investigating the effects of fusaric acid on our chasse organism.
-
How much fusaric acid are the cells exposed to?
- The phlF promoter begins responding at a concentration of 46 ug/ml5. Our cells may therefore need to be exposed to a significant inhibitory concentration before responding to fusaric acid.
Based on this literature information, each design is given the following review:
- TetR Design: This design performs worst in terms of the rate of TLP production as it expresses TLP under the weakest promoter. This design does not have a quick response time to fusaric acid, as TLP will only be expressed once TetR degrades after forty minutes.
- Antisense Design: This design expresses TLP under a promoter that is three times weaker that the T7 promoter. However, its response time to fusaric acid is dramatically faster. After about eight minutes, the antisense RNA will degrade, allowing the TLP transcript to be translated.
- T7 Design: This design expresses TLP under a significantly stronger promoter than the other designs. However, it likely has the slowest response time, as TLP production only begins once TetR degrades and T7 expresses. Furthermore, leaky expression is a big concern with this design. All the designs have the potential for leaky expression, but leaky TetR repression in this design would lead to T7 polymerase expression, and fundamentally turn on the entire expression system.
After our investigation, we conclude that the Antisense Design is “best.”

- Tegel, H., Ottosson, J., & Hober, S. (2011, January 12). Enhancing the protein production levels in Escherichia coli with a strong promoter. Retrieved October 25, 2017, from http://onlinelibrary.wiley.com/doi/10.1111/j.1742-4658.2010.07991.x/full
- Andersen, J. B., Sternberg, C., Poulsen, L. K., Bjørn, S. P., Givskov, M., & Molin, S. (1998, June). New Unstable Variants of Green Fluorescent Protein for Studies of Transient Gene Expression in Bacteria. Retrieved October 25, 2017, from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC106306/
- Philips, R. M. (n.d.). » How fast do RNAs and proteins degrade? Retrieved October 25, 2017, from http://book.bionumbers.org/how-fast-do-rnas-and-proteins-degrade/
- Bacon, C. W., Hinton, D. M., & Hinton, J. R. (2006). Growth-inhibiting effects of concentrations of fusaric acid on the growth of Bacillus mojavensis and other biocontrol Bacillus species. Retrieved October 25, 2017, from https://www.ncbi.nlm.nih.gov/pubmed/16405699
- Notz, R., Maurhofer, M., Dubach, H., Haas, D., & Defago, G. (2002). Fusaric Acid-Producing Strains of Fusarium oxysporum Alter 2,4-Diacetylphloroglucinol Biosynthetic Gene Expression in Pseudomonas fluorescens CHA0 In Vitro and in the Rhizosphere of Wheat. Applied and Environmental Microbiology,68(5), 2229-2235. Retrieved October 29, 2017.
Results
We were unfortunately unable to create our full, composite plasmid, but we did create it in three separate parts - a sensing part, an antisense RNA part, and a TLP secretion part. Our experiments involved testing the sensing and TLP export parts individually. To do this, we created a test part that combined our sensing part with a GFP reporter and another test part of constitutively expressed TLP with a histidine tag for easy isolation and detection.
Sensing Results
µTo test how our sensing system responded to the presence of fusaric acid, we created a construct with our constitutively expressed fusaric acid activated repressor (phlF), respective promoter (PphlF), and a GFP reporter (BBa_K2477008). We grew our construct in different molarities of fusaric acid (0µM, 100µM, and 500 µM) along with a negative control without a GFP reporter and a positive control with a constitutive GFP reporter. We found that sensing-reporting bacteria did not grow in any concentrations of fusaric acid, while our controls did. Therefore, we were unable to characterize our proposed sensing mechanism. Had our test worked, we expected to see decreased fluorescence with the presence of fusaric acid, with a more dramatic lowering than our positive control. We tried this test again using salicylate, another compound found to stabilize the phlF repressor, in hopes to get results of sensing. Fortunately, this compound was not toxic to the cells, but our results showed no significant decrease in fluorescence upon addition of salicylate.
TLP Expression
We planned several tests to determine the expression and activity of TLP. Our first set of tests were assays for basic TLP expression. Had these tests been successful, we planned to move on to test the ability to TLP to act as an antifungal, and ultimately the ability of our E. coli to kill F. oxysporum.
SDS-PAGE of TLP
To test expression of TLP in a bacterial cell, we created a part to constitutively expressed TLP with a 10x histidine tag (BBa_K2477006). Cells were grown overnight to full OD then sonicated. The cell lysate was purified with a Zymo Reserach His-Spin Protein Miniprep Kit.

SDS PAGE of purified protein - An SDS PAGE of the product from his-tag purification was performed. The left lane is the ladder and the right lane is eluate from the column purification, determined by nanodrop to have 12.5 ug of protein. We expected to see a band around 18 kDa but saw no significant banding at any location.
Several other SDS PAGE gels were ran (not shown) with different conditions. To account for the possibility that the TLP is insoluble, cell extract was boiled in SDS and loaded onto a gel. No significant difference in protein expression was found between the control and TLP culture.
A bacterial secretion system was created (BBa_K2477009) to attempt to secrete TLP. The culture supernatant was concentrated and loaded onto a gel along with boiled cell extract. Again, no significant banding difference could be visualized with SDS-PAGE.
From these results, we considered that the TLP expression could have been too low to be visualized on a gel. We then pursued two options--creating a new part with TLP under the control of a T7 promoter, and running a Western Blot. Due to time constraints, we were unable to assemble the T7 controlled TLP part in time for testing.
Western Blot
Using anti-His tag antibodies, we performed a Western Blot to see if our TLP was being expressed. We used a positive control of GFP with a histidine tag and a negative control of a construct without a histidine tag. We did not see any expression of TLP.
Efficacy of TLP as an antifungal
We planned to test the ability of TLP and the TLP-producing E. coli to inhibit fungal growth or cause fungal death. To do this, we initially planned to perform a disc-diffusion assay with our purified protein product and F. oxysporum and qualitatively measure difference in fungal growth between that and a control. We planned to plate the fungus and add a drop of either a purified protein product or control (positive and negative) onto filter paper around the edge of the fungal growth (for growth inhibition) and in the fungal culture (for death).
Our next step would have been to repeat this procedure, but use a cell lysate instead of protein product. This test would be to determine if our bacteria could produce enough TLP to have an effect on F. oxysporum.
Following that, our final test would be to grow E. coli with the F. oxysporum on plates and observe any fungal growth inhibition or death beyond that observed in a negative control. This test would be to determine if our bacteria had antifungal properties.

A sample of our bacteria and fungus test. The fungus was spread vertically in columns one third of the way to the center. The bacteria was spread horizontally across the center.
During our research for the fight against Fusarium oxysporum, we encountered literature that suggested the PhlF protein was weakly active without the presence of its inducer. PhlF is a repressor induced by compounds such as fusaric acid and salicylate. In our circuit, its activation triggers a TLP-producing response through a system that represses the off-switch to TLP production. Constitutive weak repressing activity causes the unnecessary metabolic strain of producing TLP in conditions without F. oxysporum. In the interest of reducing this metabolic stress placed upon our bacterial strain of choice, we decided to pursue efforts to modify this protein in order to alter its repression profile. The hope was to create a PhlF protein that would only have repressive activity in the presence of its inducer.
Typically, mutating a protein in hopes of increasing its aptitude for a particular task is difficult and likely to fail. In all likelihood, the selective pressures of evolution will have already selected for the most streamlined, functional variant(s) long ago. The PhlF protein, however, is a multifunctional repressor capable of performing a variety of tasks and responding to multiple stimuli. As such, we reasoned that it may be possible to mutate the protein in such a way that it would be unfit for use in the natural environment, but ideally suited for our particular application of it in our genetic circuit.

Cas9 screening method. (A) A target sequence is mutated in a plasmid containing an antibiotic resistance. (B) The plasmid of interest is transformed into competent cells with the Cas9/ sgRNA machinery. (C) Expression of Cas9 and its sgRNA that compliments the wild type target sequence is induced. Plasmids mutated in the target region evade Cas9 cleavage. Plasmids retaining the wild type, non-mutated sequence are cleaved by Cas9 and degraded. (D) The transformed bacteria are plated with the selected antibiotic. Cells with mutations in the region of interest maintain antibiotic resistance and survive. Cells without mutations lose their resistance and do not grow.
For the actual process of mutating the phlF gene, we set our sights on altering the operator binding region. This was a small section within the gene and to select for mutants within it we decided to adapt a side project that we had been mulling over. The goal of this project was to utilize Cas9 and an sgRNA with a base pairing region corresponding to a region of interest within a gene in order to select against non-mutants in that particular domain. This Cas9 selected mutagenesis project was originally going to be geared towards screening genomic mutants, but we elected to alter the project design to focus moreso on plasmid mutagenesis so as to increase its fitness for usage alongside our banana project.
The idea behind this project is to use the specific endonuclease activity of Cas9 as a screening tool in order to cleave genes that are unmutated in a region of interest. There are therefore two important aspects of this project that must be considered. First is the production of Cas9 and a compatible sgRNA molecule with a base pairing region corresponding to the region of interest within a gene. Second is the presence of the gene of interest that will have preferably undergone some form of random mutagenesis. These two elements would then be encoded on separate plasmids with compatible origins of replication and differing antibiotic resistance markers. Other than this, another major consideration in terms of utilizing this system would be the proper design of the sgRNA and the assurance that a protospacer adjacent motif (PAM) exists near the region of interest.
We believe that the strength of this system lies in its ability to screen for mutations that do not produce an easily observed phenotypic change. However, in order to test the efficacy of the system we opted to use GFP as it is both well characterized gene and capable of producing a variety of different mutations that would allow for easier screening down the line. The known mutations for GFP have also been well studied which made it very easy for us to identify the chromophore region of the gene as the ideal site to target with our Cas9 screening system in order to show a proof of concept.
In order to tweak our system for use alongside GFP, we first identified our region of interest within the GFP gene as the chromophore encoding region and proceeded to scan the nearby DNA for a PAM site. In the case of the Streptococcus pyogenes Cas9 that we were using, the PAM site was 5'-NGG-3'. Unfortunately, since the base pairing region for an sgRNA is limited in terms of size to about 20-25 base pairs and since there were no PAM sites near our 9 base pair region of interest, we had to improvise. We ended up making a synonymous site directed mutation within the GFP gene in order to artificially introduce a PAM site on the bottom strand. This allowed for the design of an sgRNA capable of interacting with our region of interest.

GFP targeting. GFP was targeted for Cas9 cleavage. A silent point mutation was introduced into a wild type GFP at the 183rd base pair. This mutation created a PAM sequence recognizable by Cas9. An sgRNA was designed and introduced to complement a 20 base pair segment downstream of the PAM site.
Upon constructing all of the genetic components of our project, we set about testing our system's functionality. We first transformed DH5α cells with our Cas9/sgRNA plasmid. Expression of these elements was under the control of a tetracycline promoter and the plasmid backbone was pSB1C3. The plasmid also constitutively expressed TetR, thereby allowing for the regulation of Cas9/sgRNA expression. The part number for this Cas9/sgRNA/TetR plasmid is BBa_K2477014. These cells containing the Cas9/sgRNA expression cassette were made into chemically competent cells.
The XL1-Red strain of E. Coli was used to generate GFP mutants. BBa_K2477003 (GFP (with artificial PAM site) plasmids in pSB3K3 were transformed into XL1-Red cells and cultured overnight. Part of the culture was subjected to a plasmid prep, yielding a library of mutant GFP plasmids. The rest of the culture was subcultured overnight, and the subsequent culture underwent plasmid prep, yielding another library of mutant GFP plasmids. These plasmid libraries are dubbed “Mutant GFP P1” and “Mutant GFP P2” for 1 or 2 days of passage in the XL1-red mutator strain. Alternative methods of generating mutants such as error prone PCR could have been used, but we elected to use a mutator strain due to the availability of materials and the simplicity of its use.
The resulting plasmid libraries were transformed into the Cas9 competent cells. Alongside the GFP mutant library, we also transformed our Cas9/sgRNA competent cells with a plasmid constitutively expressing non-mutagenized GFP containing a PAM site (BBa_K2477000) as well as a plasmid containing a constitutive RFP expression system. Both of these plasmid backbones were also pSB3K3. The non-mutagenized GFP served as a negative control as we expected it to be cleaved completely by our Cas9/sgRNA system. The RFP would meanwhile serve as a positive control that should not be cleaved by Cas9/sgRNA as it lacks sequence similarity to the sgRNA base pairing region.
After transformation and rescue, each culture was inoculated into LB cultures containing Kanamycin and Chloramphenicol. Each group contained cultures that were either induced, or not induced with a tetracycline analogue, anhydrotetracycline (ATC), to a final concentration of 400 ng/mL. These cultures were grown overnight and diluted by a factor of 10-5, 10-6, and 10-7 before being plated on LB agar plates with kanamycin.

LB+agar+kanamycin plates of GFP wild type uninduced (left) with ATC and induced (right) with ATC. Fewer colonies by a magnitude are seen on the plate where Cas9 expression is activated.

LB+agar+kanamycin plates of GFP Mutant P2 uninduced (left) with ATC and induced (right) with ATC. Fewer colonies by a magnitude are seen on the plate where Cas9 expression is activated.

The number of colonies on each plate were counted. Colony counts were multiplied by the dilution factor to obtain CFU/mL for each group. Error bars represent standard error between the three plates for each group.

The number of colonies on the (+) ATC plates were divided by the number of colonies on the (-) ATC with the same transformation conditions and dilution factors. Error bars represent the standard error between the three different plates for each group.
From the figures above, we generally observe a lower amount of colonies on plates where the Cas9 cassette was induced compared to when it was not induced. Of all groups, the non-mutant GFP sample has the lowest percentage of colonies in the induced group, which matches our expectations, as the non-mutant should be cleaved by the Cas9/sgRNA complex.
Once the plasmid is cleaved, kanamycin resistance is not passed down to daughter cells, thereby leading to cell death and lower culture density. The Mutant GFP P1 group did not show any significant different decrease in colony count, but the Mutant GFP P2 showed a significant decrease. We expected mutants to show an increase in colony survival due to the procurement of mutations that do not match the sgRNA base pairing region over time. However, we only see a slight increase from GFP to Mutant GFP P2. The RFP control did not have any significant decrease in colony count, but there was wide variation in the system. We expected to see no decrease in colony count because RFP is not targeted by the GFP sgRNA. It is possible that the slight difference observed between the two groups could be due to the added stress of induction, but the difference was not significant for us to conclude that Cas9 activation itself lowers colony count.
The conclusions that can be drawn from this data are limited as the distinction between the non-mutant GFP group and the other groups are not definitive. This data suggests that our system is somewhat functional, but more trials will have to be conducted for more reliable results.
Sequencing ResultsFrom the plating tests, we searched for colonies with interesting phenotypes indicative of a change in the chromophore region of GFP. We were hoping to see several different colors, or absence of color, but only ended up encountering green and white colonies. We would have preferred to run this test several times over, but were unable to complete all desired testing due to time constraints.
We selected 10 mutant GFP P1 induced colonies, 15 mutant GFP P2 induced colonies, 2 mutant GFP P1 non-induced colonies, 2 mutant GFP P2 non-induced colonies, 2 non-mutant GFP induced colonies, and 3 uninduced non-mutant GFP non-induced colonies for sequence analysis. These colonies were grown overnight in kanamycin and then miniprepped. It should be noted that only kanamycin was included in these overnight cultures in order to reduce the prevalence of the Cas9/sgRNA plasmid, while retaining the GFP plasmid. Given enough time we would have preferred to cure these cells entirely of the Cas9/sgRNA plasmid before miniprepping, but this was unfortunately not an option. These samples were then sequenced using primers specific to GFP.
Upon analyzing the sequencing data, we found that many of the colonies selected lacked mutations within the region of interest. In particular, all of the non-mutant/wild type GFP samples and all of the mutant GFP P1 samples (induced and uninduced) lacked any mutations whatsoever in the region of interest. The mutant GFP P2 induced and uninduced groups on the other hand possessed mutations at the PAM site. The PAM site that we had purposefully introduced in order to allow for sgRNA interaction with the chromophore region of the GFP gene was mutated in 1 out of 2 uninduced mutant GFP P2 samples and 7 out of 15 induced mutant GFP P2 samples. These findings are summarized in the images/charts below.

Reverse Point Mutation in GFP - A silent mutation was created with site-directed mutagenesis in a GFP protein to introduce a PAM sequence adjacent to the region we targeted. This sequence allows for successful Cas9 targeting and cleavage. After two rounds of random mutagenesis using cells from a mutator strain, a reverse mutation was observed that eliminated the PAM sequence.


PAM Site Mutation Frequency. This graph shows the percentage of sequenced samples that contained a mutation resulting in the loss of the PAM site within the GFP gene. No PAM site mutation losses were observed within the wild type/non-mutant GFP or within the mutant GFP P1 group. 1 out of 2 uninduced mutant GFP P2 samples contained a lost PAM site while 7 out of 15 induced mutant GFP P2 samples contained a lost PAM site.
From this data, we can extrapolate some interesting information. First, the Cas9/sgRNA mutagenesis method is not entirely effective in cleaving non-mutated plasmid DNA. This is demonstrated by the fact that most of the sequenced colonies were non-mutants in the region of interest. We expected that some non-mutant plasmids would escape cleavage by Cas9 simply by chance, so this finding did not come as a particular surprise.
Along with this, it can be seen that the Cas9/sgRNA system is selecting for some particular types of mutations. That is, in this case, mutations within the PAM sequence. It makes sense that Cas9 would select for mutants lacking a PAM site as it would be impossible for the Cas9/sgRNA complex to cleave this sequence. As such, plasmids losing the PAM site would escape cleavage and be propagated. Hence, there would be significant selective pressure for the loss of the PAM site. This is an issue that will need to be considered in future iterations of the project.
Additionally, comparison between the non-induced and the induced groups seems to suggest that there might be some background expression of Cas9/sgRNA despite being under the regulation of a tetracycline based promoter. Assuming that Cas9/sgRNA expression is being properly inhibited, we should see a stark difference between the number of mutants within the PAM site of the induced and uninduced samples. Of course, the sample size difference between the mutant GFP P2 induced versus uninduced groups is large (15 versus 2). Therefore, the comparable PAM site mutation rates observed in the induced and uninduced samples is not significant enough to conclude that there is leaky expression of Cas9/sgRNA.
One final conclusion that can be drawn from the data gathered is that the density of mutants within the region of interest achieved by this system is rather low. In this case, it is actually zero. A major concern in the development of this project was that the activity of Cas9 was sometimes non-specific. If significant sequence similarity is present, Cas9 is capable of cleaving off target sites. The worry here was that this would lead to point mutants in the region of interest being cleaved even though they were, in fact, mutants. To counteract this issue, we tried to obtain a high fidelity version of Cas9 which had been mutated to reduce off target cleavage. We attempted to obtain this gene from two sources, but ended up empty handed in the end. We were forced to settle for using a normal fidelity Cas9 gene provided to us by a professor on campus. This could potentially be the reason why we see a lack of mutations in the region of interest. Of course, the lack of mutants could also be due to the fact that not enough colonies were screened. Ideally, we would've preferred to screen many times more colonies than we actually did, but due to time constraints and limited resources, this was not an option.
Overall, the data collected for the Cas9/sgRNA mutant screening system seems to indicate that it is somewhat effective. The reduced culture density of non-mutant/wild type GFP with induction seems to suggest that non-mutant plasmids are being cut to some degree. Additionally, the prevalence of PAM site destroying mutations within the mutagenized GFP samples supports the notion that the screening system is selecting for plasmids that can avoid cleavage. While we had hoped that these cleavage avoiding plasmids would contain mutations within the region of interest rather than at the PAM site, the findings presented here at least point to the potential for this system to be used in mutant screening. Incorporating a higher fidelity Cas9 gene into our system could greatly improve its effectiveness as a whole.
Future PlansGoing forward, we would like to continue to characterize our Cas9 based mutant screening system. We plan to collect more data on its implementation and effectiveness and would like to demonstrate that it can be used in lab to effectively aid in screening mutants. We also plan to make efforts towards obtaining a higher fidelity version of Cas9 for incorporation into our mutagenesis selection system. Comparing the effectiveness of this enzyme against the effectiveness of the normal fidelity variant could help to show that the system effectiveness increases with increasing Cas9 fidelity.
Our end goal for this project is to standardize the system such that it can be utilized by other laboratories as a tool to screen for mutants. We would like to develop sets of step by step procedures in order to guide iGEM teams through the entire process of mutating a protein in the region(s) of interest and screening with our Cas9 system. The hope is that we will be able to produce a simple and well characterized mutagenesis system that can be used to further the endeavors of future iGEM projects.

UMaryland iGEM has focused its hardware project efforts to bringing low cost DIY lab equipment to expand synthetic biology to community labs, high schools, and developing countries. We have successfully built a thermocycler and an ultra low -80 C freezer, and this year we are expanding on this effort.
The UMaryland iGEM Lab-in-a-Box is a compilation of various lab hardware that allows synthetic biology research and education in settings with limited resources. It comprises of a microcentrifuge, incubator, shaker, and vortex machines in one box that is open source, compact, and customizable. While these parts will cost thousands of dollars to purchase, it can be built for under $300. The construction of the box requires no soldering, drilling, and uses all off-the-shelf and 3D printed parts to function, making construction and repair easy. With the lab-in-a-box and lab equipment found in typical high school classrooms, transformations, overnight cultures, and minipreps can be conducted, with further possible modifications to increase its functionality.
We aimed for this project to be used primarily in high schools, where there may be limited resources. In addition, it presents an interdisciplinary learning opportunity, since the physics, engineering, and biological aspects of the box can be taught and explored. When parts inevitably break of malfunction, the students can learn how to troubleshoot and repair the appropriate machinery. It is also modular and expandable, with ideas for further expanding the ability of the box given, so that high school students can add components that they need instead of purchasing extra equipment.

The included guide to assembly includes detailed explanations of the principles behind the box, how it is achieved, a full parts list, and assembly instructions including pictures of almost every step. It also includes ideas for expansion and a usage guide. We've used feedback from our outreach efforts in order to make the guide more clear for high school audiences.
The guide can be download at https://static.igem.org/mediawiki/2017/3/3f/T--UMaryland--Hardware_Guide.pdf
The 3D rotor files can be downloaded at https://static.igem.org/mediawiki/2017/9/9d/T--UMaryland--Hardware_CAD.zip in both .stl and .ipt formats.
The Arduino script can be downloaded at https://static.igem.org/mediawiki/2017/7/7c/T--UMaryland--Hardware_Script.zip

There are four main components of the box: the motor and speed controller unit, the Peltier plate assembly, the 3D printed rotors, and the Arduino microcontroller.
Motor and speed controller
The motor (Golden Power A2212-10 1400KV Brushless Motor) is a DC brushless motor that provides the shaking, centrifugation, and vortexing functions. It runs on DC 7.4 - 11.1 V, and provides 1400 RPM/V (rotations per minute per volt). The maximum speed depends on the voltage provided to the motor. This motor was chosen for its high maximum speed and pre-soldered bullet connectors, which allows easy connection to the speed controller. It draws a maximum of 16 amps and 180 W of mechanical power, making it powerful enough to drive the centrifuge (see modeling page for more detail).

The electronic speed controller (ESC - Velotech Magic Multirotor 30A Electronic Speed Controller with BEC) provides two functions: controls the motor and provides a 5 V output for the rest of the electronic assembly. The ESC comes with pre-soldered female bullet connectors, matching the rotor. It accepts a PWM input (pulse width modulation) - which means short repeated bursts of voltages, from the Arduino and translates it to differential rotational speed of the motor. It has a 2A BEC, providing a 5V 2A output for the Arduino and H-bridge components that uses a 5V input. When programmed, it can provide braking (using active current to stop the motor), reverse mode, and more.
Peltier plate assembly
A thermoelectric cooler kit was used due to its low cost and effectiveness. It is prebuilt to not require drilling of screw holes and allows for secure mounting of both heatsinks. It is a complete kit with heatsink, thermal paste, Peltier plate, and 12V DC fans, making it ideal for our use. However, we opted for better quality thermal paste and a higher power Peltier plate to meet our needs.
The TEC1-12712 Thermoelectric Cooler was used for our Peltier plate, which was chosen because it allows more current compared to the one that comes with the kit. A Peltier plate works by turning electrical work into a heat differential, with a hot side and a cold side. The difference in temperature is determined by voltage, current, and the temperature of the hot side. More voltage and higher current will result in a bigger difference between the hot side and cold side, but that means more heat is generated and has to be removed from the hot side of the plate for the cold side to be cold.
For example, even if the temperature difference is 80 C because more power (voltage times current) was put into the system, if the hot side is 100 C due to the lack of efficient cooling of the Peltier, the cold side would only be 20 C. However, even if there is only a 40 C temperature difference with less power, if the hot side is maintained at 20 C, the cold side would be at -20 C. Therefore, it's a balance of the heat able to be dissipated by the hot side and the power supplied that achieves the coldest temperature.
We opted for air cooling because it is easier to maintain and more cost-effective versus water cooling solutions. However, this makes it hard for us to model heat dissipation and since air cooling is not as effective as water cooling, the lowest temperature reached was only around 8 C.

The Peltier plate assembly is controlled by an H-bridge, which allows both a modulation of the voltage and the direction of current. It accepts a PWM input and a directional input from the Arduino, allowing us to control the temperature difference created by the Peltier plate in response to the temperature in the box. The PWM input controls the voltage, and therefore the power given to the Peltier plate. The more input (from a higher PWM) is given, the more power will be delivered, leading to a larger temperature difference between the hot and cold sides of the Peltier plate.
It also has the ability to change the direction of the current based on input from the Arduino, so that electrons are flowing in the opposite direction. This allows the cold side of the Peltier to become the hot side and vice versa. Therefore the Lab-in-a-Box can act as both an incubator and a chilled centrifuge depending on the application.

The temperature in the box is sensed by a thermsister, which is a resistor that chances its resistance based on the temperature. It is an NTC (Negative Temperature Coefficient) probe, meaning that the resistance increases as temperature decrease. In the Lab-in-a-Box, the resistor is wired in series with a 10 kOhm resistor, and the difference in voltage reflected by the change in resistance (following Ohm's Law - V = IR) is sensed by the Arduino
3D Printed Rotors
Three rotors are designed for the Lab-in-a-Box, the centrifuge, shaker, and vortex function. The centrifuge and shaker parts were designed with capacity in mind - we did not want to limit high schools or labs by the small capacity when making overnight cultures or doing minipreps. Each rotor is mounted in a #10 Nylon lock hex nut, which fits with the threaded adaptor that comes with the motor. A Nylon lock hex nut was used for two reason - the increase height of the nut provided a larger surface area for the glue joining the rotor and the nut, and the nylon lock prevented the rotors from becoming stuck on the motor.
The design of the centrifuge and vortex pieces were relatively straightforward - the centrifuge rotor relies on perfect balance of forces while the rotor is spinning while the vortex piece relies on rapid spinning of a slightly off centered rotor. The designs were obtained and modified from commercial equipment.
The shaker rotor was more complex to design because the fundamental method of shaking had to be different from a commercial shaker. A commercial shaker uses an unbalanced weight that spins around a motor, with a platform on top that is elastically held to the weight. Therefore when the weight spins, it generates a shaking motion that is translated into the circular movement of the platform. The platform where the samples lie is only loosly held to the unbalanced weight, therefore the movement of the samples is translational rather than rotational.
In our design, the shaker relies on rotational motion rather than translational motion. While some shakers rely on rotational motion, they simply spin the samples 360 degrees up-side down to achieve the shaking effect. This is not feasible with overnight cultures because the container is open to allow air flow, and will leak if flipped. We achieved the shaking effect by using an unbalanced force that is at an angle. When the motor spins, it provides an inherent shaking motion, but since the samples lay at an angle, the force experience by the side closer and further from the axis of rotation will be slightly different - leading to a slight vortexing effect.
We also had to overcome the lower limit of the speed of the motor. The lowest possible speed is still around 2000 RPM, 10x what is normally needed. Therefore a manual pulsed spinning of the shaker was necessary. The program would activate the motor for a short time, wait until the shaker has come close to a stop, then spin again. This spun the shaker at a lower rate.
Arduino
An Arduino Mega was used to control and provide feedback throughout the system. It accepts input from the temperature sensor and the push-buttons on the LCD screen, and outputs to the ESC, H-Bridge, and the LCD screen. The Mega was chosen due to its faster processing speed and memory capacity, which was beneficial for our program and makes it more able to be expanded in the future. However, it can be run from a lower powered Arduino such as an Arduino Uno if necessary.

The user interfaces with the Lab-in-a-Box through a 16 x 2 character LCD screen. It has the buttons for up, down, left, right, select, and rest (rst) that allows control of the various functions such as setting the incubator temperature and centrifuge speed.
Before purchasing a motor, we needed to calculate to see what the requirements for power would be.
First we calculated the maximumload (weight) of the rotor plus the samples with the parameters:
- Weight of microcentrifuge tube: 1 g
- Capacity of microcentrifuge tube: 1.7 mL
- Density of LB: approximately equal to water, 1 g / mL
- Capacity of rotor: 30 tubes
- Density of PLA: 1.25 g / cm3
- Volume of rotor (given by AutoDesk Inventor): 106.706 cm3

The maximum load on the motor will be 214.38 grams
Next, we determined the desired speed, which is determined by the miniprep protocol. We proved that minipreps need only 10,000 x g to be effective (see results). Converting RCF (x g) to RPM, with the radius (r) of our rotor being 60 mm:

To find the required torque, we set our desired angular acceleration of the centrifuge to 42 rad/s, which translates to reaching our max speed in 30 seconds. To calculate the moment of inertia, we modeled the centrifuge rotor as a hoop, where the mass of the rotor itself is negligible compared to the total mass of the samples. Ignoring friction and aerodynamic drag:

We found the required torque is 0.0324 N m, which we will use to calculate the RPM generated with this torque.
We found the technical data on the motor in the spec sheet and found the maximum power of the motor to be 180 Watts. However, the motor cannot run at max power for a long period of time due to heat constraints. The max-efficiency current, which lessens the heat constraints is 12A, where the efficiency is about 75%. The efficiency is defined by the ratio of mechanical power (used to drive rotational motion) to the electrical power (current times voltage). At 12 A and 11.1 V (which we will use to approximate the 12 V of our system, we have an efficiency of about 75%. This means electrical work (12 A * 12 V) is 144 W, which will generate 0.75 * 144 = 108 W of mechanical work.
Using the DC Motor power equations given by Micromo we calculated the maximum RPM from the given torque.

Using the calculated torque to drive the load and theoretical maximum power given, the model predicts that the motor generate an RPM much more than the 12,199 RPM necessary. In reality, the motor will draw less current once it hits the max rotational speed of 1400 * 12 V, rather than reach this theoretical maximum. This proves that the motor is capable of running the centrifuge at a speed fast enough for miniprepping plasmid DNA.
Stress SimulationThere were two aspects of the lab-in-a-box that we wanted to simulate: the stress on the PLA based centrifuge rotor during centrifugation and the effect of unbalanced shaking on the motor. This was extremely important for safety, as we didn't want the 3D printed pieces or the motor shaft to be destroyed and cause injuries in the process. We use AutoDesk Fusion and PTC Creo in order to simulate the stress put on them.
For the centrifuge, two tests were simulated. The first was using AutoDesk Fusion, where the centrifuge piece was subject to the angular acceleration found through our calculations. Using PLA for our rotor piece and plastic casing filled with dense metal as our microcentrifuge tubes, we found that there was minimal concern for too much load, but that the load was mostly concentrated in a ring between the holes where the centrifuge tubes were placed. We increased the distance between the holes for centrifuge tubes as a result, even though it decreased our capacity.

For our shaker, we used PTC Creo and subjected the parts under 100 N m of torque, which represents the initial stress felt as the shaker was accelerating. We found that there was managable stress placed on the motor shaft, which allowed up to proceed with our 3D printing and construction
When designing a product, it's important to define a problem or need correctly before finding the solution. During our design process, we realized we never questioned whether or not the speed (17,900 x g) required in the miniprep protocol was actually required for plasmid extraction. If it were possible that it could be done at a lower speed, it would both reduce cost, improve durability, and have a safer product. Given the same starting culture, we tested the effect of centrifugation speed throughout all steps of the miniprep protocol to determine the speed of the motor in our lab-in-a-box. A commercial benchtop microcentrifuge was used to perform this miniprep, and a Qiagen miniprep kit was used for plasmid DNA extraction.


Our results show that DNA recovery is independent of centrifugation speed between 17.9k and 8k x g RCF. The plasmid DNA concentration was similar throughout all speeds, and the gel confirmed that the plasmid was indeed the constitutive RFP vector (~2 kb). However, the 8k x g RCF bands on the gel looked more faint in two thirds of the samples, and we decided to pursue a speed of around 10,000 x g RCF for our design.
Centrifuge CalibrationThe centrifuge needs to be calibrated prior to use because the input from the Arduino is in units of microseconds rather than RPM. Since the motor does not have a way to sense this, we needed to correlate the microseconds sent to the speed controller to the speed generated.
We used an infrared reflectance sensor to calibrate the centrifuge. The sensor sends infrared light, and detects how much of it has bounced back. The body of the motor is reflective, so we placed a thin strip of electrical tape on the motor, which will absorb all of the infrared light. From this, the Arduino can detect the time elapsed between two readings when infrared light has been absorbed, and therefore can calculate the speed of the motor. The calibration was conducted with the centrifuge rotor placed onto the motor.

From our results, we found that the speed of the Arduino could not be fast enough to detect very high RPMs. which would need sensitivities of tenths of millisecond. We were able to get three clear data points to correlate the control action / microseconds sent to the ESC and the corresponding RPM. From the maximum control action, we extrapolated to about 12,000 RPMs, which corresponds to 11,290 x g RCF, more than enough for miniprepping plasmid DNA.
Incubator TestThe incubator needs to be calibrated as well because the thermsister can only cause a change in voltage. While a theoretical approach can be used to calculate the temperature, we decided to do an experimental calibration instead, due to the sensitivity of samples to temperature. Three temperature points were obtained by comparing the analog input with the temperature in the box. This was input into the program to create a standard curve.
 Shaker Test
Shaker Test
The shaker was tested using a constituively RFP expressing system. A starter culture was incubated and split into three categories: positive control - a commercial incubator shaker, experimental group - our lab-in-a-box shaker, and negative control - our lab-in-a-box incubated without shaking. A n of 3 was used per group.


Immediately after taking the samples out of the shakers, it was clear that the samples lacked strong RFP expression like the positive control. However, the turbidity was greater than the negative control, and there was expression seen in one of the experimental samples. Pelleting of the cells resulted in a clear decrease in pellet size and red fluorescence compared to the positive control but still much better than the negative control.

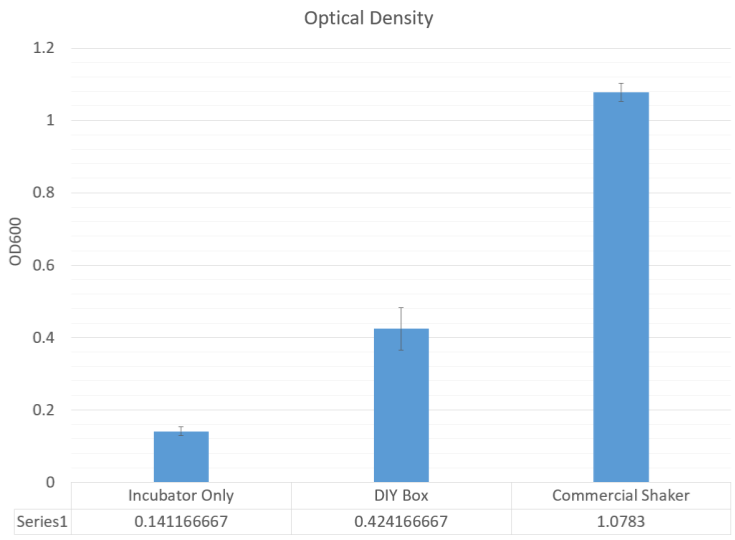
Measuring the RFP fluorescence and the optical density, it was clear that the cells did not fluoresce at all compared to the commercial shaker, but still had half of the optical density. This shows that while there was growth, it was not in log phase, when the promoter of the RFP cassette would have been activated.
We miniprepped the cultures in a commercial centrifuge to test if the optical density may have been due to contamination, which could have increased optical density but would not have the plasmid DNA that matched our RFP expression vector. We found that plasmid DNA recovery proportionally matched the optical density.

While our shaker did not result in strong RFP expression and log phase growth, we believe that other methods of induction such as IPTG would cause fluorescence because it does not depend on log phase growth. It would also be beneficial for replication of plasmid DNA for sequencing and other assembly procedures.
We have demonstrated that:
- The miniprep protocol requires only 10,000 RCF to isolate plasmid DNA effectively.
- The lab-in-a-box centrifuge can reach up to 12,000 RPM, generating 11,290 RCF
- The thermsister responds linearly to changes in temperature to maintain a constant temperature.
- The shaker can induce some expression of genes under a log phase promoter, and grow culture with around 50% plasmid DNA recovery.
To understand the context of the problem of DIY hardware, we first approached the Baltimore Underground Science Space (BUGSS) which is a community lab in Baltimore, MD. We wanted to know if DIY hardware is something that community labs are in need of, and if the type of equipment that we are designing were in need. We were told that due to our proximity to research laboratories and federal institutions such as the National Institutes of Health, that basic equipment that we were building were not a top priority.
We refocused our efforts towards an educational audience, where basic equipment are more in need. We had two audiences in mind: a high school iGEM team and an AP biology classroom with synthetic biology instructions. After writing our guide, we wanted to get feedback on the machine and get suggestions on clarity of the guide.
We visited Broadneck High School in Annapolis, MD, where we were able to introduce iGEM and synthetic biology to the Science Honor Society there. Then, given all the disassembled materials and the guide, they were told to try and build the machine again without guidance from us. We received valuable pointers on what parts were not clear, and the need to include techniques such as how to strip a wire. We incorporated this feedback into our guide. However, in under an hour and a few pointers, they were able to get a functional motor running.
Safety was a top priority during the construction of this project. A high speed centrifuge can become very dangerous if not handled properly. Since we are aiming our project towards high school students, it is especially important that we ensure the safety of the students using this box. We use controls such as software limits and safety built into the design itself in order to ensure proper use.

The centrifuge is the most conerning part of the project, due to its very high speed. When unbalanced and centrifuged for an extended period of time, the rotor can warp and break. We've extensively tested what occurs under unbalanced conditions and found that the point of failure is the glue holding the nut onto the rotor itself. During unbalanced motion or high heat from extended use, the glue will break or become loose, forcing the rotor off of the motor. This results in just the nut spinning on the motor by itself. The rotor also cannot penetrate or severely damage the styrofoam box even when unbalanced, only causing several nick, making the user safe from possible accidents. We've also implemented a software limit on the time of centrifugation to 5 minutes in order to prevent warping or breakage.
The Peltier plate can also pose a burn hazard if left heating uncontrolled. There are two mechanisms implemented to prevent this: there is a software limit of 50 C on the heat inside the incubator. Due to uneven ventillation, the heatsink attached to the Peltier will be hotter, but this will prevent any major burns from occuring. In addition, the H-bridge controlling current to the Peltier plate is by default off, and only turns on when required. The H-bridge shuts off current to the Peltier after the incubation or shaking periods are over, resulting in rapid cooldown of the heatsink.
Over the past few years, the UMaryland iGEM team has taken particular interest in making synthetic biology more accessible to the general public, especially to high school students. This has been demonstrated in our previous efforts in 2015 to develop a DIY thermocycler made from common household appliances like a hair dryer and in 2016 to create a DIY ultra-low freezer. Our hardware project this competition, a lab-in-a-box containing many pieces of equipment necessary to molecular cloning, also works towards this goal of increasing accessibility to synthetic biology.
This year, we wanted to take our efforts one step further than finding low cost solutions to lab equipment availability. We envisioned creating an educational program capable of teaching an audience about iGEM and the basics of synthetic biology in a way that would be both meaningful and memorable. In prior years we have taught lessons at local high schools, but they often seemed lacking in terms of highlighting how useful and exciting synthetic biology can be. Speaking with the students after these lessons showed that they only remembered a small portion of the material presented. This was very disappointing to us as we wanted our audience to gain meaningful knowledge from our lessons, so this year we did things a little bit differently.

Sample of material presented - This image depicts some of the hand drawn pictures created by our team in order to help teach students about synthetic biology. The watercolors are meant to be eye catching and more engaging for students than looking at typical plasmid diagrams generated using sequence design software.
First off, we decided to develop a project solely for the purpose of enriching our outreach to high schools. This project involved demonstrating synthetic biology's usefulness via the implementation of simple metal detection genetic circuits into our lessons. These circuits served well for our purposes in multiple ways. They were simple and easy to explain in terms of the genetics, they could produce a visible result via fluorescent reporter genes, and they had the potential to solve real world problems. Specifically, these metal detection circuits could be easily related back to the Flint, Michigan water crisis. Being able to underscore the ways in which synthetic biology could be used to solve actual issues was a must for us, so the metal detection project was ideal for incorporation into our high school lessons.
Along with developing a project that would garner the attention of students and act as the focal point of our in class presentation, we also worked hard to revise the way in which we teach our lessons. Since students were struggling to retain information from our lectures, we did some research and decided to make lesson plans based on the principles of active learning. This meant doing things such as clearly stating learning objectives prior to and after the lesson, having class wide and small group discussion, and engaging students with thought provoking questions. All of this culminated in a lesson that encouraged participation and effectively showcased the need for synthetic biology as well as some of the underlying scientific principles. Utilizing this lesson plan, the high school lectures that we led were very successful and can be read about further in the outreach section of the website.
In order to use these metal detection circuits in our lessons, we first had to design, construct, and characterize them. We decided to do this for the detection of three different types of metal: copper, lead, and zinc. Planning out the genetic circuits was relatively simple as they only required a few components. First was a constitutively expressed metal sensitive repressor, second was a promoter regulated by said repressor, and third was a fluorescent reporter gene under the control of the metal regulated promoter. In the case of our copper biosensor, the genes/elements involved include the CueR repressor, the pCopA promoter, and an RFP reporter gene. Similar systems were designed for the lead and zinc biosensors using the PbrR and ZntR systems. The fluorescent proteins used for these circuits were YFP and GFP respectively.

Copper detection circuit A: Copper biosensor in the presence of copper ions Copper ions inhibit the binding of CueR to the pCopA promoter, allowing for expression of RFP. B: Copper biosensor in the absence of copper ions In the absence of copper ions, CueR binds to the pCopA promoter and inhibits RFP expression. A reduced amount of RFP is synthesized due to some leaky expression of the RFP gene.
Upon completing our designs and assembling our plasmids, the next step was proving that they worked. Because this was not our main project and since we were trying to effectively characterize of our parts prior to our the start of our high school visits, we decided to focus testing solely on the copper biosensor.
The first set of tests conducted involved inducing RFP expression using varying concentrations of copper in overnight cultures. Glycerol stocks of XL1-Blue cells transformed with the copper detection plasmid were used to inoculate 4 culture tubes containing 5 mL of LB broth. The appropriate antibiotic was added and then the cultures were induced to a final copper (II) sulfate concentration of 0 µM, 5 µM, 100 µM, or 500 µM. After incubation with shaking overnight, these cultures were spun down in a centrifuge and compared qualitatively. Clear differences in the color of the cell pellets could be seen between the 0 µM, 100 µM, and 500 µM groups. The 5 µM group appeared to be about the same coloration as the 0 µM group.

Copper sensing overnight results - XL1-Blue cells containing the copper biosensor were incubated overnight with varying concentrations of copper. The different copper concentrations induced varying levels of RFP expression. This can clearly be seen in the different coloration of the pelleted cell samples.
Following this qualitative proof of functionality, we proceeded to perform a more quantitative test. XL1-Blue cells containing the copper biosensor were grown overnight without induction to maximum culture density. Approximated 1 mL of this culture was then used to inoculate 20 mL of fresh LB in a 50 mL Falcon tube. This new culture was grown at 37 C with shaking until it reached an OD600 above .4. 1 mL samples of this culture were then removed and induced with copper (II) sulfate to final concentrations of 0 µM, 25 µM, 50 µM, 100, µM, 250 µM, 500 µM, and 1,000 µM. Aliquots of each induced sample were then loaded into 5 wells of a 96 well plate. This plate was placed into a shaking/incubating plate reader set at 37 C and measured for fluorescence (584 nm excitation 612 nm emission) every 5 minutes for 180 minutes in total. OD600 readings were taken at the beginning and end of this time course and a linear regression was used to approximate the OD600 for each time point. The OD600 data was used to normalize the fluorescence data in order to generate the graph below.

Copper induced RFP expression over time - XL1-Blue cells containing the copper biosensor plasmid were grown to log phase and induced with varying concentrations of copper. Fluorescence was then measured every 5 minutes thereafter and normalized against OD600.
This graph shows clear differences in terms of the expression profiles of the different experimental groups. While the cells induced with 25 µM and 50 µM copper show little to no difference from the non-induced sample, the other induction groups show a clear upward progression in terms of fluorescence against increasing copper concentration. Overall, this figure as well as the qualitative testing show that our copper biosensor can reliably detect copper concentrations greater than or equal to 100 uM.
From our research and after visiting the Washington Suburban Sanitation Commission Patuxent Water Treatment Plant, we found that normal drinking water typically contains around 0.01 mg/L copper. The EPA limit set on copper in drinking water is meanwhile 1.3 mg/L. When converted to µM, typical drinking water copper levels are approximately 0.2 µM whereas the EPA limit is set at approximately 20 µM. Since our biosensor sensitivity is limited to 100 µM, detection of copper levels above the EPA limit would require water samples to be concentrated prior to any form of induction. While this would not be the most convenient in terms of actually testing water for copper contamination, the functionality of this part has served our team well in terms of helping to educate high school students and the next generation of synthetic biologists.
Going forward, our team is looking to characterize our zinc and lead detection systems that were designed but not tested. Assuming that these systems function similarly to the copper biosensor, we will assemble all three detection systems into a single, master metal detection plasmid. Incorporating the design of this plasmid into our future lessons would to show students one extra way in which synthetic biology can be utilized.

Along with developing this master plasmid, we plan to spend time refining our lesson designs. We got a lot of really useful feedback from our first high school visits and we hope to incorporate the comments/critiques that we received into any future high school outreach efforts. We expect that future UMaryland iGEM teams will continue to use this lesson plan as a basis for engaging the community and teaching students about the wonders of synthetic biology.
iGEM projects reside in their context, and the purpose of human practices is to learn more about the problem and the situation that surrounds the problem. This requires a multi-disciplinary approach that involves discussing our project with stakeholders that are from a variety of backgrounds. To understand the potential impact of our project, we contacted multiple individuals in a wide variety of fields and asked for their feedback and their possible concerns.

From the business perspective, we contacted Beyond the Peel, an organization that aims to empower independent banana farmers in Ecuador and Peru, and to import fair trade organic bananas. We were told that the lack of Panama disease in South America meant that farmers are more concerned about local diseases that are currently afflicting the banana plant more than a disease localized in Asia and Africa. These diseases, along with the daily concerns of the weather and pests, are the farmer’s primary concerns, although we were told that if we had a good product that would protect the farmers, and if the disease has begun to manifest in the Americas, there would be no backlash against using foreign or GMO solutions.
In addition, we learned that there are many reasons beyond the technical challenges why the Cavendish banana is the major crop being imported. This mainly is due to infrastructure challenges, since shipping times, routes, and distribution mechanisms all depend on the Cavendish, whose ripening properties are well known. In addition, consumer demand for bananas that aren’t the Cavendish is miniscule, and no investments on building the infrastructure for different types of bananas that may be more resistant to Panama Disease, isn’t being made. We also spoke to the United Nations Food and Agriculture Organization (UN FAO) about our project and received the perspective of a policy administrator. We found that there was a World Banana Forum hosted by the UN FAO that aims to tackle labor, disease, and agricultural issues through a collaboration of industry, academia, and governments. From our conversations with those not in the scientific community, we found that biosafety and biocontainment was a major concern, which included the potential pathogenicity of our genetically engineered bacteria.


We also spoke with Dr. Juan Robalino, a researcher tackling the problem of Panama Disease, about our project, and we discussed the regulatory hurdles of conducting synthetic biology overseas. He is from Ecuador, and he founded / is the CEO of the startup Cronicas, which aims to develop crops that are more resistent to virulent and fungal infections. As a researcher in genetically modified bananas, he was able to explain that outside of the US, the precautionary principle with regards to recombinant DNA is applied to even research in the lab, requiring a permit which may take years. Since bananas are not affected in the Americas, research has to be conducted in Southeast Asia, which is both the major producer of bananas consumed outside the Americas and the country most devastated by the disease. The governments of the Southeast Asia also view this as an industry issue that companies need to solve, and that governments do not need to intervene on this issue. Currently, there are tissue banks (which are used to culture and grow the banana plants) in Taiwan that are working on finding resistant varieties. He also notified us that there isn’t much of a push towards solving this crisis because banana markets are depressed, meaning there is overproduction and a very low cost of bananas. He forecasted that once bananas become a more valuable crop, there will be a more concerted effort.
To understand the regulatory hurdles, if they were to be tested in the US first, we spoke with Dr. Eric Olson and Dr. Carrie McMahon of the Federal Drug Administration and Dr. Chris Wozniack of the Environmental Protection Agency, the two organizations that would regulate the use of our system. We learned that Bacillus amyloliquefaciens has been used as a fungicide in the past, and that thaumatin, which is similar to TLP, has preliminary evidence to be safe for ingestion by humans. We also learned that the FDA wouldn’t regulate our system because it does not directly impact the food and that the US Department of Agriculture would not regulate our system because we are not engineering plants, leaving only the EPA as the sole regulator. Although the regulation of genetically engineered bacteria versus a symbiont is more strictly controlled, there is enough precedence to consider our project more feasible compared to the engineering of banana plants themselves.

As a team, we noticed that the synthetic biology portion of the high school biology curriculum was often skipped due to various reasons, chief among these being a lack of resources. As a result, we wanted to create a metal detection system that could be used by high school teachers as a hands-on experience to teach their students about synthetic biology. While we were unable to actually bring this detection system to the high schools due to safety restrictions, we were still able to talk to these students about iGEM, synthetic biology and its applications, basics of plasmid assembly, and basics of protein expression through a workshop that utilized active learning principles.
For this workshop, we visited two local high schools, Rockville and Hammond High School, in central Maryland. We first visited Rockville High School, where we were presented with a small group of high school freshmen, who had a relatively limited exposure to biology. As a result, we had to modify our original presentation slightly to tailor it more to our audience. Instead of talking as much about synthetic biology, we focused more on the basics of biology, such as explaining the central dogma. By engaging these students in activities that employed the subject material we were covering, it was clear that by the end of the workshop, they understood the basic principles behind plasmid assembly and protein expression.
The next high school we visited was Hammond High School, where our audience was a larger group of mostly juniors and seniors, who had previous exposure to some basic biology concepts. As a result, we were able to use our originally created presentation. Because this was a bigger audience, we were able to break the students off into groups. This allowed one member of our team to facilitate discussion with and ask some questions about the content to the smaller group before we pulled the groups back together and continued with our workshop.
It was extremely rewarding to see these students be so engaged and interested in synthetic biology. Despite their minimal exposure to it, they were able to pick up the principles and basics of the subject extremely quickly. The students from Hammond High School even sent the team a thank-you note after the workshop.
One of the challenges facing synthetic biology is the negative stigma surroungind genetically engineered or modified organisms. Therefore, it's important that we reach out to the community as a part of iGEM to discuss synthetic biology and advocate for their use by presenting a case where synthetic biology can be used to solve an issue.
As a result of talking with Ravdeep from Beyond the Peel, we found that one of the reasons why the Cavendish is the most popular banana is due to the lack of consumer demand. If people are not willing to buy these bananas, there will be no need for an infrastructure to be built around it. We were also interested to see if there was another banana candidate that would be able to replace the Cavendish, and if people would buy it as a result. Genetic diversity is important in protecting the population of Cavendish bananas. Crops that are more resistant will prevent the fungi from spreading more rapidly, and isolate populations where there the disease is present.

We found from specialty grocery stores four other types of bananas that are available to be purchased, and we had participants test them out. We had bought in addition to the Cavendish: burro, manzano, thai, and red bananas. They were all ready to be eaten when ripe, which is different from the cooking bananas such as plantains. They have different flavor profiles and textures compared to the Cavendish. The flavor profiles of each banana are determined by the starch, sugar, and nutritional composition. The bitter, dry taste of unripe banana is due to the high starch content of the non-Cavendish bananas. Starch is also responsible for the crunch / hard taste. When ripening, starch breaks down into the sugars fructose, sucrose, and glucose, which give bananas their sweet taste and creamy texture. Red bananas in particular have a different color and taste due to their higher beta carotene (Vitamin A) content. Cooking the bananas (akin to plantains) helps to break down the starch.

We asked the following questions:
- How do each type of bananas taste? How does it compare to the Cavendish?
- If ripening the bananas mean that they turn completely black, would you eat a black banana?
- Would you buy one if you saw them in the grocery store?
- Some of the bananas are dual-purpose, bringing out a different flavor profile when cooked. How do you feel about cooking bananas?
- Did you know there were other varieties of bananas beyond the Cavendish?
- Do you shop with diversity in crops in mind? Do you buy organic, non-GMO, etc.?
We found a variety of responses to the event, a lot of participants liked the red bananas and its more complex sweet taste to be better than the Cavendish. Some liked the more tart flavors of the other bananas but it was split between the red bananas and the Cavendish. We found that the majority of participants have never heard of bananas that weren’t the Cavendish, and they mentioned that the would start looking for this in the stores.
We also talked about the project and synthetic biology, and how we can apply synthetic biology to the problem of the disappearing Cavendish banana. They were interested in learning more about it, and they preferred that we used a soil probiotic rather than genetically modifying bananas themselves.

One of our continued collaborators this year was BUGSS, the Baltimore Underground Science Space. They are a community lab based in Baltimore that provides anyone in the area interested in synthetic biology with the resources to design and conduct their own experiments.
We reached out to them in the beginning of the summer initially regarding our hardware project. Because community labs often have limited resources, we were curious to see if they had any insight into what specific equipment we should include in our lab-in-a-box. During this visit, we received feedback regarding our hardware project, learned about what it was like to work in a community lab, and were invited to come back to visit and to collaborate with BUGSS’s own high school iGEM team.

We visited BUGSS again in early October for its Open-House event. There, we presented the community with a basic description of our project and asked them to taste the Cavendish, burro, manzano, thai, and red bananas. As found at our Farmer’s Market visit, most people preferred the Cavendish and red bananas, finding that the other varieties were too bitter or starchy. In addition to this, we were able to listen to BUGSS’s high school iGEM team give a presentation on their project regarding plastic degradation and provide feedback and suggestions.

Our final visit was later in October when we were invited back to present on our projects to local synthetic biology enthusiasts. They found our projects to be very exciting and gave us feedback on the presentation formatting and content.

We went to the Patuxent Water Filtration (WSSC) in Laurel, Maryland. The purpose of this visit was to obtain an industrial outlook on methods of water purification and the deleterious effects of metal contamination in drinking water. In light of the events that occurred in Flint, Michigan with widespread lead poisoning, it was emphasized to us the importance of pre-emptive corrosion treatment in pipelines (using small amounts of orthophosphate).
From this visit, we were introduced to the various steps in the water filtration process beginning with flocculation, followed by sedimentation, granular filtration (anthracite coal), and UV sterilization for cryptosporidium. Similarly, the implication of our metal detection project is to give high school students an idea of what the entire water filtration process entails, why metal detection matters on a large scale and how metal detection plays a critical role in the purification of water. Using what we learned from this visit to the WSSC, we hope to integrate a component for metal detection in water into the overall lesson plans.

We also learned about the effects of front-end chlorination during the filtration process which leave trihalomethanes (THM) byproducts in the water, components that were suspected to be carcinogens.
Manganese dioxide coating on filtration systems to prevent corrosion, but manganese leeching causes discoloration in water. While the water may be clean (according to the standards in place), from the business perspective, consumers will not want to purchase this water simply because it is not visually appealing. This is a problem for filtration plants because in order to introduce a manganese dioxide coating into the system, they would need to shut off the water to that system and allow for the coating to form. This coating takes upwards of 7 days to form, which is an issue for business.
UMaryland iGEM is made of dedicated undergraduates who are passionate about improving the world through synthetic biology. They are from a variety of backgrounds, majors, and interest that contribute their talents through lab work, outreach, fundraising, wiki coding, video editing, social media, and photography. They dedicate their time through a spring seminar, summer work, and finishing up of the project in the fall.

|
Annie Trang Annie is a junior studying Neurobiology & Physiology major. She is interested in pursuing a career in the medical field. In addition to wet-lab work, she is involved with both the team’s high school education outreach and art work. Otherwise, she can be found discussing ethics, pursuing creative projects or working with physicians in an urgent care clinic. |
||

|
Asha Kodan Asha is a rising sophomore who is majoring in Biology with a minor in Religious Studies. This is her first year on the UMaryland iGEM team! In addition to her passion for synthetic biology, she is an avid writer and opinion columnist for the Diamondback Newspaper. She also helps with behavioral inhibition research at UMD's Child Development Lab. During her free time she enjoys traveling and watching true crime documentaries. |
||

|
Cameron Harner Cameron is a senior in Bioengineering. After graduating he plans to work as a consultant in the healthcare industry. He joined iGEM to get hands on lab experience and work on a project with real world applications. Outside of lab he enjoys sports, game of thrones, and just about anything outdoors. |
||

|
Chun Mun Loke Chun Mun is majoring in Biochemistry and Cell Biology/Genetics. He is currently applying to MSTP programs, and hopes to pursue a career in medical research. Outside of class, Chun Mun enjoys playing music, watching Netflix, and sleeping. |
||

|
Cynthia Uzoukwu Cynthia is a senior with a double major in bioengineering and criminal justice. After she graduates, she hopes to go on to medical school to become a neurosurgeon. She joined iGEM because she likes that students get to choose their own research topic, based on their analysis of what real life problems synthetic biology could help combat. Apart from science, she likes anything involving criminal law. |
||

|
Deven Appel Deven is a junior bioengineering major on the pre-medical track. With a desire to explore synthetic biology, he found iGEM had the perfect goals for him. iGEM also provided a first-hand look at how these principles of biology and engineering could be applied to actual, beneficial goals in the world, and he was happy to see such a passionate, determined team around him at the University of Maryland. Deven is also the president of an a cappella group and assists with a biophysics lab on campus. |
||

|
Jacob Premo Jacob is a Junior Biochemistry and Microbiology double major. He is particularly interested in studying genetics and the mechanisms underlying drug effectiveness. As a second year team member, Jacob has enjoyed assisting with new member training and managing the laboratory space. Outside of iGEM, Jacob spends his time reading, watching movies, and playing with his dog. |
||

|
Joyce Song Joyce is a sophomore neurobiology & physiology major. She joined iGEM because she loved the concept of a student-ran research team that tackles real world issues with synthetic biology. In her free time, she loves to go play with her dog, go on hikes, and binge-watch Netflix. |
||

|
Paula Kleyman Paula is a junior studying bioengineering. This is her second year on the UMaryland iGEM team. She really enjoyed working on the team the year before and learning how to design and create a novel synthetic biology project and came back for more. She is also interested in pursuing a career in education, possibly finding a way to integrate her biology and engineering knowledge with classroom work. Her favorite activities outside of academics are running, singing, and playing guitar (but not at the same time). |
||

|
Richa Beher Richa is a sophomore Cell Biology and Genetics major at the University of Maryland who aspires to become a physician-scientist. She joined iGEM to gain research experience and explore synthetic biology applications with like-mided individuals. On campus, she is part of service organizations, Terrapin Trail Club, and the Club Triathlon team! In her free time, she enjoys eating good food, backpacking, listening to music, and hanging out with friends. |
||

|
Rohith Battina Rohith is a sophomore biochemistry major at UMD. He is interested in pursuing a career in the field of infectious diseases research. He enjoys doing iGEM because of its application to real world problems and how it fosters teamwork. In his spare time, he plays tennis and reads books. |
||

|
SangHo Jee SangHo is a junior biochemistry and cell biology major at UMD. He coded the wiki, led the hardware project, and led the outreach efforts of the banana project. He is interested in synthetic biology of eukaroytic systems, and is pushing for UMaryland iGEM to move beyond single E. coli to thinking about interacting populations and engineering yeast. |
||

|
Seth Cohen Seth is a junior biochemistry major at the University of Maryland. Outside of science his favorite things to do are play basketball and write fiction. His favorite food is probably falafel on pita bread with tomatoes, cucumbers, feta, lettuce, tahini, and hummus. His favorite animal is definitely the elephant. Another kind of interesting fact about him is that he is vegetarian. That's pretty much all you need to know about Seth! |
||

|
Thea Orstein Thea Orstein is a rising Junior at the University of Maryland, College Park, majoring in Bioengineering with a concentration in Biotechnology and Therapeutics Engineering. After graduating from UMD, She plans to pursue a PhD in Biophysics or Molecular Biology. This summer, she is working as a lab member and as a project developer on the UMaryland iGEM team. In her free time she enjoys running, hiking, and hanging out with friends. |
||

|
Vaidehi Bhagat Vaidehi is a rising junior in the University Honors Program at the University of Maryland, majoring in Bioengineering on the Therapeutics Engineering and Biotechnology track. She is a researcher on the UMaryland iGEM team and aids in the various projects that are being developed. She hopes to enter the biopharmaceutical industry after graduation and then eventually pursue a Master's Degree. In her free time she enjoys relaxing, enjoying new foods, and dancing. |
||

|
Yuzhu Shi Yuzhu is a sophomore biochemistry and microbiology major. She loves doing research and thought that iGEM would be a great way to further pursue that interest. Her other hobbies include trying out new foods and restaurants, going to the gym, and hanging out with friends. |
||
|
|
|
UMaryland iGEM Advisors
UMaryland iGEM is lead by two faculty advisers: Dr. Edward Eisenstein in the Fischell Department of Bioengineering, and Dr. Jason Kahn in the Department of Chemistry and Biochemistry. We were founded with the help of Dr. Boots Quimby, former Assistant Director of the Integrated Life Sciences Honors Program at the University of Maryland, College Park. They help evaluate project proposals, attend regular lab meetings, connect us to resources on and off campus.

|
Dr. Jason Kahn Dr. Jason D. Kahn is a biophysical chemist who studies protein-nucleic acid interaction and engineering. He is best known for studies of DNA looping, bending, twisting, and cyclization, as well as hybridization thermodynamics for modified bases. He teaches a variety of chemistry, biochemistry, and molecular biology courses, which he credits for initiating his interest in synthetic biology. Dr. Kahn was a graduate student at UC Berkeley and a post-doc at Yale before coming to Maryland in 1994. |

|
Dr. Edward Eisenstein Dr. Edward Eisenstein is a Fellow in the Institute for Bioscience and Biotechnology Research and an Associate Professor in the Fischell Department of Bioengineering at the University of Maryland. Trained in modern structural enzymology, his current research interests are focused on protein and biosystem engineering for discovery and application in plants and microorganisms. |

|
Dr. Booth "Boots" Quimby Dr. Quimby is the former Associate Director of the Integrated Life Sciences honors program in the Honors College at the University of Maryland. Prior to joining the Department of Cell Biology and Molecular Genetics at UMD as a full-time instructor, she earned her Master's of Arts in teaching from the University of South Carolina, after which she taught high school science in Atlanta, Georgia for eight years. She then returned to graduate school and received her doctorate in genetics and molecular biology from Emory University. |

|
Naren Bhokisham Naren is a Graduate Student in Molecular and Cell Biology at the University of Maryland, College Park. He received his undergraduate degree in Industrial Biotechnology from St. Joseph’s College of Engineering, Anna University, India. He works in the intersection of synthetic biology, metabolic engineering and biomaterials, involving assembly of enzyme cascades on various interfaces to generate small molecules and engineering microbes to display novel phenotypes in response to small molecules. Apart from science, his pursuits include traveling, running and latin dancing. |
We at UMaryland iGEM would like to acknowledge the following sponsors for their generous financial and material support

We would also like to thank the generous individual donors on our Experiment.com and our UMD Launch crowdfunding campaigns.
Special Individuals
We would like to acknowledge and thank following people for providing us with the resources, advice, and connections that made our project possible
- Dr. Angus Murphy, UMD Plant Sciences - Provided valuable advice and aided in securing funding and lab space for the team.
- Ravdeep, Beyond the Peel - Provided valuable information on the outlook of banana farmers.
- Dr. Juan Robalino, Cronicas
- Dr. Eric Olson, Federal Drug Administration
- Dr. Carrie McMahon, FDA
- Dr. Chris Wozniack, Environmental Protection Agency
- Dr. Caroline Nguyen, Washington Suburban Sanitary Commission
- Nicole Horvath, WSSC
- Teresa Moore, UMD Bioengineering - Helped in the management of our funds.
- Ron Noble, UMD Bioengineering - Aided in the ordering and receiving of materials.
- Catherine Carroll, UMD Bioengineering
- Catherine Carroll, UMD Bioengineering
- Fabiola Mijares, UMD CMNS
- Cierra Clinkscales, UMD IBBR
- Naren Bhokisham, UMD Bioengineering graduate student - Provided us with advice on whatever we needed help with. This included a lot of advice on equipment usage.
- Justin Sylvers, UMD Bioengineering - Highlighted UMaryland iGEM in The Catalyst
- Dr. Jason Kahn, UMD Biochemistry - Acted as a mentor to the team. Attended lab meetings, provided advice, organized an iGEM seminar, and helped to make iGEM a wonderful experience.
- Dr. Edward Eisenstein, UMD IBBR - Acted as a mentor to the team. Attended lab meetings, provided advice, organized an iGEM seminar, and helped to make iGEM a wonderful experience.
- Dr. Boots Quimby, UNC Chapel Hill - Helped to establish the team in 2014. Served as a mentor and aided in managing finances.
iGEM Teams and Collaborators
We would like to acknowledge the students and advisors of the following groups, who contributed to the designs of our projects, and enabled us to learn through outreach efforts.
- Baltimore Underground Science Space, Baltimore, MD - They provided suggestions to improve our lab-in-a-box project.
- 2017 iGEM Team, College of William and Mary, Williamsburg VA - They collaborated with us in order to test our copper biosensor.
- 2017 iGEM Team, University of Virginia, Charlottesville VA - They hosted us at the Mid-Atlantic iGEM meetup.
- Rockville High School, Rockville, MD - They acted as the first group to listen to our synthetic biology lesson.
- Hammond High School, Columbia, MD - They invited us to teach our synthetic biology lesson at their school.
- Broadneck High School, Annapolis, MD - Their students had a chance to test out and give feedback on the hardware project
- 2015 iGEM Team, University of Glasgow, Scotland, UK - They provided us with sequences and information the phlF system and adapting it to E. coli.
- 2014 iGEM Team, Wageningen University, Netherlands - We used their information to come up with our sensing design and learn more about another synthetic biology approach to combat F. oxysporum
- 2011 iGEM Team, Unicamp-EMSE, Brazil - They provided us information and parts for protein secretion.
Laboratories
We would like to thank the following groups for their scientific advice and support.
- Joseph Roberts Lab, UMD Department of Plant Science and Landscape Architecture - They provided us with space in their laboratory in order to conduct our work
- William E. Bentley Lab, UMD Department of Bioengineering - They allowed us to use their equipment including their shaker/incubator, nanodrop, and plate reader
- Eisenstein Research Group, UMD IBBR - They served as willing mentors for us and completed the PCR reaction and subsequent Gibson Assembly to isolate the Cas9 gene that we struggled to clone for some time. They also provided us with a His-tag antibody.
- Kahn Research Group, UMD Department of Chemistry and Biochemistry - They served as willing mentors for us, provided us with a Cas9 plasmid, and provided us with samples of several reagents.
- Wang Research Group, UMD Department of Chemistry and Biochemistry - They aided in performing all of our sonication and SDS-PAGEs for us. An iGEM team member working in this lab conducted the procedures.
- Volker Briken Lab, UMD Department of Cell Biology and Molecular Genetics - They assisted us in running a western blot. An iGEM team member working in this lab conducted the procedure.
All of the cloning, assembly, and wet lab work not mentioned above were conducted by the iGEM students
Donors and Sponsors
We would like to thank the following groups for their generous contributions, either financially or materially.
- Department of Plant Sciences and Landscape Architecture, UMD
- Fischell Department of Bioengineering, University of Maryland College Park (UMD)
- A. James Clark School of Engineering, UMD
- College of Computer, Mathematical, and Natural Sciences, UMD
- Integrated Life Sciences, UMD Honors College
- Institute for Bioscience and Biotechnology Research, UMD, the National Institute of Standards and Technology
- iGEM Foundation, Cambridge MA
- Qiagen
- Integrated DNA Technologies
- New England Biolabs
- Agilent Technologies
Wiki
The wiki was coded from scratch using the following plugins:
- jQuery
- panZoom - allow drag and drop scrolling
- Pace - loading screen
- Animate.css - bouncing animations
The font is Raleway obtained from Google Web Fonts. We would like to acknowledge W3Schools and StackOverflow as valuable references. No templates were used to code this wiki.
| Registry No. | Part Name | Description | Link |
| BBa_K2477000 | iGEM GFP (E0040) mutant with added PAM site | The GFP gene was originally obtained from the iGEM 2017 DNA kit. Site directed mutagenesis was used to create a synonymous mutation at base pair 186 where a cytosine was incorporated into the top strand to replace a thymine. This acted to create a PAM site for Cas9 from S. pyogenes (5' NGG 3') on the bottom strand. This mutation was made in order to allow for the design of an sgRNA capable of interacting with the chromophore encoding region of GFP. The biobrick number of the sgRNA designed by our team for this purpose is BBa_K2477001. | http://parts.igem.org/Part:BBa_K2477000 |
| BBa_K2477004 | Cas9 | This is the Cas9 gene that was utilized in our experiments. It is capable of cleaving specific DNA sequences when expressed alongside an sgRNA. This part was taken from addgene plasmid pML107. | http://parts.igem.org/Part:BBa_K2477004 |
| Registry No. | Part Name | Description | Link |
| BBa_K2477003 | GFP with PAM site (BBa_K2477000) expression system | This part constituitively expresses GFP. The synonymous mutation introducing a PAM site in the GFP gene allows for the interaction of this part with an sgRNA overlapping the chromophore region of GFP (BBa_K2477001). This allows for selection against non-mutants in this overlap region when expressed alongside the sgRNA and a Cas9 nuclease. | http://parts.igem.org/Part:BBa_K2477003 |
| BBa_K2477006 | Rice TLP With 10x His-tag Expression System | Strong constitutive expression of rice antifungal thaumatin-like protein. To be used for protein extraction. | http://parts.igem.org/Part:BBa_K2477006 |
| BBa_K2477008 | GFP based fusaric acid biosensor | This part includes constitutive production of the phlF repressor. The repressive ability of this regulator is stabilized by fusaric acid and salicylate. The PhlF protein inhibits the immediately downstream promoter which regulates the transcription of GFP. A decrease in GFP production should therefore accompanies the presence of fusaric acid/salicylate. | http://parts.igem.org/Part:BBa_K2477008 |
| BBa_K2477010 | TLP Secretion System | This circuit constitutively expresses the antifungal agent, TLP, tagged with a 6x His-tag, a 2x GS linker, and an HlyA secretion peptide. The HlyA tag allows for secretion using the HlyB/HlyD system. Both HlyB and HlyD are also encoded on this part and are under control of an arabinose promoter. Please note that there is no scar site between the end of TLP (the GSGS linker) and the start of the HlyA tag. | http://parts.igem.org/Part:BBa_K2477010 |
| BBa_K2477013 | Copper Biosensor | This part constitutively expresses the CueR repressor. This repressor then inhibits the expression of RFP from the downstream PcopA promoter. In the presence of copper, the repression of CueR is inhibited leading to greater RFP expression. Fluorescence of this construct has been shown to increase with increasing copper concentration in both a qualitative and quantitative manner. | http://parts.igem.org/Part:BBa_K2477013 |
| BBa_K2477014 | Cas9/sgRNA GFP Mutant Selection System | This part contains the machinery to express Cas9 and an sgRNA targeting the chromophore region of GFP (E0040). The expression of both components of this system are under control of tetracycline promoters. The part also encodes for constitutive expression of the TetR repressor. This thereby allows for tight regulation of Cas9 and sgRNA expression for controlled mutant selection. This part is meant for use alongside a GFP variant containing an artificially induced PAM site (BBa_K2477003). | http://parts.igem.org/Part:BBa_K2477014 |
Random mutagenesis is a valuable technique because it produces novel protein structures with potentially useful functions. Unfortunately, global mutagenesis operations also generate a vast array of fruitless changes. In our project we define potentially useful mutations as mutations that occur within a small region of interest in a gene. To determine the ratio of potentially-useful-mutations to definitely-useless-mutations produced by a global mutagenesis operation, we take a dive into probability theory.

Consider the following scenario:
- We grow a colony of cells from a single cell.
- At each cell division, there is a probability p that the cell will mutate desirably.
- The cell divides d number of times before being mini-prepped.
Our goal is to find the density of cells that have mutated desirably at the end of d divisions. To do so, we find the probability that any given cell has mutated desirably. Finding this probability is tricky because desirable mutations propagate throughout generations. We must therefore find the probability that any given cell in the final generation is NOT desirably mutated. The graphic below more readily demonstrates the math involved to analyze the cell population at the end of d divisions.
If the probability that any given cell is NOT desirably mutated is (1-p)d, the probability that any given cell IS desirably mutated is 1-(1-p)d. Rather than being concerned with cell mutations, we remember that we are truly interested in mutations of individual bases. Multiplying 1-(1-p)d by the size of our region, r, yields the probability that any given cell contains a potentially useful mutation.
We reach the following result:

Where M is the density of cells that have potentially useful mutations.
This model assumes that:
- Cas9 always cuts the on-target site;
- any off-target cuts are inconsequential;
- all mutations are non-lethal.
The following describes a realistic application of our project:
Using XL-1 Red cells, 1 in 1,000,000 bases is mutated at every cell division.1 We grow a colony of XL-1 Red cells for 30 generations, and are investigating a plasmid region that’s 20 bases large.
 Therefore 0.06% of plasmids will have a selectable mutation at the end of the mutagenesis operation
Therefore 0.06% of plasmids will have a selectable mutation at the end of the mutagenesis operation
At the end of 30 divisions, 1 cell will have become over half a billion. By implementing Cas9 screening on our globally mutagenized cells, we will transform a mixture of only 300,000 genetically unique circularized plasmids. Each of these plasmids has mutations in the region of interest.
Furthermore, finding the partial derivatives of our expression for M gives us a better idea of how this technique works. The following equations demonstrate the relative importance of each variable.



where M is the potentially-useful-mutant density, p is the mutation rate, d is the number of divisions, and r is the target region size.
Since p is usually on the order of one one-millionth; d is usually on the order of twenty or thirty; and r is usually twenty, we can see the typical contribution of changing each term. For coarse adjustment of the M value, the mutation rate should be altered. For fine adjustment of the M value, the generation number d should be altered. This foresight is useful for synthetic biologists who want to plan their lab work around the results of the mutagenesis operation.
Thus, our concise mathematical model and its derivatives are a guide for how to use the project. They inform the synthetic biologist about the variables that will impact their success with the technique, and they tell the synthetic biologist how to change those variables in order to ensure a better outcome.
1. Competent Cells For Random Mutagenesis - Details & Specifications. (n.d.). Retrieved October 29, 2017, from http://www.genomics.agilent.com/article.jsp?crumbAction=push&pageId=648
Enforcing and emphasizing laboratory safety is a crucial component in the iGEM framework and in the greater scientific community. This year the University of Maryland iGEM team abided by safety measures appropriate for biosafety level 1 as our project involves the engineering of several common E.coli strains (DH5-Alpha, BL21, BL21 DE3) to inhibit the spore germination and mycelial cell growth of a non-pathogenic strain of Fusariuam oxysporum.
Safe project designA main priority of the team was to not only combat panama disease and protect the banana from extinction, but to also design an environmentally safe solution that would not involve harmful soil treatments such as fumigation. To accomplish this, we created a system that would take advantage of safe, naturally derived antifungals. If our project was eventually applied to Banana fields, several modifications would be implemented. First, instead of using E.coli as we did for proof of concept, Bacillus amyloliquefaciens would be chosen as the chassis because of its soil compatibility. Moreover, the target of our transgenic bacteria would be the pathogenic F. oxysporum, the perpetrator of panama disease. In terms of our metal sequestration project, which was designed for outreach applications, the University of Maryland’s department for environmental safety was consulted for proper metal (copper, lead and zinc) disposal.
Safe lab workTo commit to the lab safety lifestyle, we enforce strict lab policies and practices ranging from waste disposal to lab techniques. Before the beginning of the summer term, each lab member completed Chemical Hygiene training and Biohazard safety training through UMD’s department of environmental safety and an additional, specific iGEM training session lead by the lab coordinator, Jacob Premo. During this training several important lab protocols were taught such as, transformation, colony picking, bacterial culturing, plasmid DNA extraction, gel electrophoresis, and waste disposal (autoclave). Appropriate lab attire was emphasized, which requires long pants, closed toed shoes, and protective eyewear at all times in the lab.
Although our team is only working with BSL1 organisms, live cultures are frequently used. To reduce the risk of contamination, team members are required to wear nitrile gloves when handling samples, work near a flame when easily contaminated samples are open and autoclave all liquid and solid waste often.
May 30 - June 1
School’s out for summer! Time to relax, enjoy the summer sun, take a swim, and -oh yeah- get working in the lab. We started this short post Memorial Day week with fully organizing and setting up the lab. Moving on to our wet lab work, we transformed competent cells with parts from the kit distribution that would be necessary for our projects. The parts we needed were pTet (BBa_R0040), TetR (BBa_C0040), and a strong Anderson promoter (BBa_J23100). We also created stocks of the pSB1C3 and pSB1A3 backbones by amplifying the linearized DNA also provided in the kit with PCR.
Unfortunately, only the transformations of the TetR was successful. We moved on to making overnights of these cells and extracting the plasmid DNA with a miniprep kit. We checked our progress for the week by running a gel with the amplified linear backbones and digests of the transformed plasmid DNA.
And... Success!
June 5 - June 9
Our first full week and what a scramble - the new members were forced to fend for themselves as the returning ones weren’t able to come in as periodically - it’s ok though, they managed. This week we attempted transformations of the pTet and Anderson promoter parts, this time only being successful with the pTet (later on to find out that the Anderson promoter had a different antibiotic resistance than we thought). We also combined the TetR piece we had transformed last week with a double terminator piece (BBa_B0015) we had from last year. The successfully transformed cells were prepared as overnights and then later miniprepped. We then performed a 3A assembly of the pTet piece with the TetR/Double Terminator construct, transformed into competent cells, prepared overnights, and miniprepped the DNA. We also restocked our supply of agar plates. From this week’s work we have assembled this piece:
June 12- June 16
We finally got the Anderson promoter transformed and miniprepped! We also put in a ribosome binding site (BBa_B0034) at the beginning of last week’s construct using PCR reactions with primers that included the RBS sequence and inserted the product into a pSB1A3 backbone via Gibson Assembly. However, when we tried to verify this construct (both through running a gel and sequencing) we found something had failed. Suspecting there could have been an issue with the backbone we used, we tried to make more backbone stocks (pSB1A3, pSB1C3, and pSB1K3) with PCR amplification. We also transformed more plasmids from the kits into competent cells. These parts are to be used for the metal sequestration project as reporters. The parts we transformed were RFP (BBa_K518012), YFP (BBa_E0430), GFP (BBa_E0240 and BBa_I13504 ) and CFP (BBa_E0420). These parts are an assembly of a ribosome binding site, a fluorescence gene, and a double terminator. We also transformed a strong ribosome binding site part. Then we prepared overnights and did minipreps.
June 19- June 23
Ready, set, glow! With our projects being put on pause as we awaited the arrival of gblock, we decided to spend the days working on our contribution to the InterLab Study
Once our TLP gblock came in for our Banana Project, we attached it to an A3 backbone. We also received samples of a nonpathogenic strain of Fusarium oxysporum and streaked it onto our homemade potato dextrose agar plates.
Since our PCR insertion of an RBS failed, we decided to use a different approach, the 3A assembly method, to attach the RBS in front of our TetR/TT/pTet construct. Now we have this:
June 26- July 7
We tried to attach an Anderson promoter to the beginning of the TetR construct we had been working on and and RBS to the end with 3A assembly, but both kept failing. The issues were likely caused by the relative small sizes of the parts we were adding. We added a constitutive strong Anderson promoter to the TLP piece we ordered to be later used for observation of the efficacy of expressed TLP.
July 10 - July 14
We received our Cas9 project gblocks - two halves of Cas9 and an sgRNA, and performed Gibson assemblies to insert them into our backbones (the two Cas9 pieces were designed to be combined together with the backbone in a 3-part Gibson and the sgRNA was designed to combine with a backbone). The Gibson of the sgRNA was successful but the Cas9 Gibson was not. We also attempted to add on to our TetR construct by using PCR to add on an Anderson promoter in the front. We decided we would use GFP as our gene of interest that would be selected for with mutations, so we used site-directed mutagenesis to introduce a PAM sequence recognizable by the Cas9 into the chromophore region of the GFP. For our Banana project, we needed to create agar plates that would sustain both the Fusarium oxysporum and E. coli, so we tested out different combinations of potato dextrose (for the fungus) and LB (for the E. coli) and found a 0.5x solution of both worked best. We also received our phlF gblock (the sensing part), inserted it into a backbone using Gibson, and transformed it. We got a new antibiotic this week too! We created our Kanamycin stock and made some agar plates with Kanamycin.
Our progress on the TetR regulatory part:
July 17 - July 21
We tried again at our Cas9 Gibson assembly, but again were not successful. Our many trials of this Gibson kept failing, so we devised new ways to take action. We also tried to attach an RBS at the end of our TetR part using 3A assembly. Unfortunately this too did not work. Our gblocks for the metal detecting parts came and we successfully used Gibson assemblies to attach two of them into a backbone, the third did not successfully assemble. These plasmids are meant to make E. coli fluoresce different colors in the presence of Zinc, Copper, and Lead (one fore each). We also had our first round of testing TLP expression and function. We plated our TLP producing bacteria with the fungus and looked for zones of inhibition. As it goes, we found it did not work. We figured a disc diffusion assay with extracted TLP would be our next step.
July 24 - July 28
We ran into the same issues as we had before with our Cas9 project. The Gibson of Cas9 was not working - our gels were not producing the bands we anticipated. Additionally, we were not able to attach and RBS to the end of our TetR construct using a 3A assembly. On the bright side, we were able to extract a histidine tagged protein (which we hope is TLP) from our TLP expressing E. coli. We also attempted a 3A assembly of our phlF sensing part with a GFP marker to be used for testing. And our individual metal detecting plasmids were completed!
July 31 - August 4
The new month brought a new wet lab schedule, with some members leaving and some joining. It was also prime vacation time so we were a bit short handed but we survived…. barely. Unfortunately, this week brought many difficulties. We were having the same issues with our Cas9 project as before, our gel imaging system was not working, and there were problems with sequencing, so we were not able to make much progress this week. We decided to add a secretion part to our Banana project design and found a part in the kit that we transformed into E. coli to attach at the end of our TLP. We also made agar plates with varying concentrations of fusaric acid to test what concentrations of this toxin would cause our cells to die. We also started working on our complete Metal project plasmid and did a 3A of our lead and zinc plasmids.
August 7- August 13
This week, our Experiment.com page to fund our iGEM team got approved. We did some work with the banana project by ligating some different parts together and miniprepping. We also ran a fluorescence test to check for the RFP to make sure it was functional. Since fusaric acid killed some of the E. coli, we are testing its toxicity to normal competent cells as well. We are almost finished with the metal plasmids and should be able to start testing soon.
August 14 - August 20
This week, we were really trying to focus on the banana project since it has encountered a number of unexpected roadblocks and has been lagging behind some of our other projects. We attempted to do some ligations to attach a secretory component and checked it by running it on a gel. We attempted to use this new plasmid with an even stronger promoter in fusaric acid. Unfortunately, all concentrations of fusaric acid killed the E. coli and only the control group grew. We may have to look at a different way to test our project since fusaric acid is not allowing us to check for phenotypic changes.
August 21 - August 27
For the banana project, we worked on adding a secretory system in for the TLP. Our metal plasmids are all ready to be tested, but we need approval to test the lead plasmid since lead is a hazardous material. We started reaching out to local high schools that might be interestested in having us pay them a visit. We were running experiments with our hardware project to determine how fast our centrifuge will need to be to be feasible for most protocols.
August 28 - September 3
It’s the first week back to school for a lot of us which gave us very little time to do some lab work. We attempted to assemble Cas9 once again. However, there was an issue with contamination so the experiment was invalid. We started talking about doing a writeup on the modeling that we have been doing alongside our projects. With the competition approaching, we are beginning to look for events that allow us to talk about our project and get some experience presenting.
September 4 - September 10
This week signaled the second week back to school for team. We attempted to use Gibson assembly for the Cas9 plasmid into an A3 and a C3 backbone, but it was unsuccessful. We also attempted to purify TLP using a His-tag miniprep kit, but it did not yield any significant results. Now that the semester has started, we anticipate that lab work might slow down due to time constraints.
September 11 - September 23
We made cryostocks of all three of our metal plasmids. We also completed our TLP+HLYABC plasmid. Unfortunately, our Gibson mix seems to lack efficiency so we decided to purchase a new one. We also were not successful in attaching an RBS to pAND+RBS+TetR+TT+pTet. This week, we were making lesson plans for some of our anticipated high school visits.
September 18 - September 24
This weekend, we went to a Young Researchers conference and explained our project through a poster. We noticed some flaws in our presentation and our poster and are working on fixing it. We did some preliminary shaker testing and incubation tests for our hardware project. We attempted to ligate Cas9 into a C3 backbone and a T7 promoter with RFP. We ran the Cas9 on a gel, but it was unsuccessful. We started looking at different methods to raise funds for our team and we have been planning some of our high school visits to teach students about synthetic biology.
September 25 - October 1
This week, we attempted to ligate the Cas9 with a C3 backbone using Gibson assembly, but it was unsuccessful. We performed some basic fluorescence testing with the copper and zinc plasmids. There wasn't much difference between the induced and uninduced samples of the zinc sample. There was some RFP fluorescence from the copper plasmid when induced in a 100 uM concentration of copper. For the banana project, we attempted to ligate the T7 promoter to TLP, but it was not successful. There was no detectable TLP on the gel from the TLP+Hly plasmid. We attempted to run a copper fluorescence assay, but the procedure was performed incorrectly so the data had to be thrown out. We tested the phIF controlled GFP expression with salicylate since it has similar effects to fusaric acid ( without killing the cells). However, the results weren't significantly different between the induced and uninduced samples so we will need to redo the experiment.
October 2- October 8
This week, we attempted to ligate the TLP with our promoter and ligate the Copper plasmid with the Lead and Zinc plasmids. Unfortunately, the TLP ligation was unsuccessful. We miniprepped the copper plasmid to check to make sure that it was completed. We tested the phIF+GFP part with salicylate. We received samples from William and Mary from their project to test and we sent them our copper plasmid. We made some new agar plates in preparation for their samples. Unfortunately, our fluorescence testing was delayed due to factors out of our control so we didn’t get to complete it this week. We also went to another nearby high school to talk to them about synthetic biology and introduce them to the concepts. This weekend is the Farmer’s Market and another meeting with BUGSS. We hope to talk to farmers about some of the work we did in relation to the banana project. We are also starting to put things together onto the website and compiling our research from the summer. Our LaunchUMD page was confirmed to launch soon to help us fundraise more money.
October 9- October 14
We sequenced some of our plasmids this week using GeneWiz. We also started producing overnights for our constitutive TLP and normal competent cells to look for the expression of TLP using a Western blot. Once again, we tried to ligate the T7 promoter with the TLP plasmid. We made overnights of the cas9-RFP plasmid. We didn’t have a lab meeting this week because many of our members were out visiting the WSSC. We received a positive inducible control of TLP as well as a conjugated antibody for the Western blot so it got pushed back until the next week.
October 16- October 21
We finally tested out the copper part using a fluorescence test and it is functional. We also completed the tests on the plasmids that we received from William and Mary. We were also able to isolate the Cas9 part and it is also very close to completion, it's just missing one final part and has some small mutations. We also ran our Western blot this week. Unfortunately, it was not successful and we didn't see any expression from the blot. We had another two high school visits during the week and they both went well and we talked about synthetic biology and plasmids and iGEM itself. We have another collaboration with BUGSS this weekend as well.
October 22- October 27
This week, we ran a gel on the final Cas9 samples to find one that was correctly completed to submit. The gel for the Cas9 looked good, but the sequencing did not, so we are attempting to redo it before the deadline. We didn't end up doing anything else for the banana project. Now that the year is wrapping up, we are submitting all of our parts before the deadline and making the final touches to the website.
Interlab Study
Expansion of synthetic biology requires collaborative work, where scientists are able to both share and reproduce constructs. However, the lack of a standard protocol for fluorescence measurements prevents direct comparison of data between teams. To propose a solution, the 2017 iGEM’s Fourth International Interlab study invited teams to contribute data towards establishing a standard measurement of fluorescence of fluorescein.. The 2017 UMaryland iGEM team has performed the interlab study with the plate reader protocol and transformation of the specified parts as directed by the Measurement Committee’s guidelines.
Procedures
Absorbance measurements were collected using a n of 3
All measurements were performed with a standard 96 well flat-bottom plate, with each well filled to a total volume of 100uL for every solution tested.
Calibration of our microplate reader used LUDOX-S40 as a single point reference to appropriately convert absorbance values into a standard OD600 measurement. Absorbances were measured for four replicates per LUDOX and deionized water at 600 nm.
Determination of the fluorescein concentration required a standard curve. A fluorescein 1x stock solution of 50uM was prepared via resuspension of 2x solution (100uM) in 1mL of 1xPBS followed by a another addition of 1mL 1xPBS solution. Four replicates of 1:2 serial dilutions were completed for a row of 11 wells, one of which contained only the prepared 1x stock. Measurements were performed at 600nm with our standard GFP settings (excitation: 485nm, emission: 538nm).
Following our standard curve, cell measurements for fluorescence were tested with varying devices in E. coli K-12 DH5-α. After transformation of the plasmids into the DH5-α cells, two colonies were selected per device and inoculated in 5mL of LB medium with Chloramphenicol. The cells grew overnight for approximately 16 hours at 37 C with 220 rpm shaking. The OD600 measurement of the overnight cultures were performed immediately following the time span. Cultures were then diluted to a target OD600 of 0.02 in 12mL of LB medium and chloramphenicol in a 50mL Falcon tube prior to incubation. 500uL were retrieved from the cultures at time 0, 2, 4, and 6 hours of incubation for absorbance and fluorescence readings on the 96 well plate (consisting of 4 replicates per colony of a test device), resulting in a total of 16 sample measurements per replicate.
All measurements were inputted into the standard iGEM InterLab 2017 excel document for respective calculations.
Results
Microplate Reader
The microplate reader calibration indicated a correction factor of 5.556 to convert absorbance into the corresponding OD600 measurement.
Standard Curve
Figure 1. Standard curve for fluorescein fluorescence measurements via 1:2 10 serial dilutions.

Figure 2. Standard curve presented on a log scale for fluorescein fluorescence measurements via 1:2 10 serial dilutions.


Our results confirm how the increase in the fluorescein concentration increases fluorescence of the cells over time. To compare the data, we reviewed the promoters and ribosome binding sites of each test device. While Test Devices 1-3 maintained the same ribosome binding site (B0032), each differed in the following promoters listed from weakest to strongest: J23101, J23106 and J23117 for 1-3 respectively. The absorbance results suggest that J23106 reflects about 25 times greater absorbance J23101 when paired with B0032. Similarly, J23117 is approximately 35 times greater in absorbance compared to J23101. After comparing to promoters, Test Devices 4-6 keep the J346100 RBS constant to compare the promoters. The listed from weakest to strongest include: J23101, J23106 and J23117 for 4-6 respectively. The absorbance graph demonstrates J23101 low level of fluorescence (with one colony demonstrating slightly higher fluorescence than expected) compared to both 5 and 6. While J23106 demonstrates 40 times the strength of J23101, J23117 indicates a slightly stronger promoter with approximately 42 times the strength of J23101.
Further review of the mean fluorescence over time that most of the test devices demonstrate sufficient fluorescence between three to four hours. Following that time span, the florescence begins to decrease. For devices with the J23106 promoter, fluorescence begins to decrease past the two hour mark.
In comparing the relative strengths of the promoters in combination with the RBS, the difference between the fluorescence measurements may be attributed to the strength of the RBS thus affecting the level of fluorescence (where J346100 may be stronger than the B0032). With data from other teams participating in the Interlab study, the UMaryland team hopes that the collection of all fluorescence data will better help with the standardization of fluorescence to account for varying promoter and RBS strengths.
Collaboration
College of William and Mary
BackgroundWhile attending the Mid-Atlantic iGEM meetup, we got the chance to speak with the William and Mary team about their efforts to control gene expression speed. We thought that the idea to use a non-endogenous protease/protein degradation tag system in order to establish reliable control over the rate at which genes are expressed was a really interesting idea. We ended up discussing how our teams could provide assistance to one another and came to the agreement to collaborate on several fronts.
Testing by UMarylandOur team took on the task of testing several constructs sent to us by William and Mary. In total, they sent us six plasmids termed A through F. Plasmids A-E consisted of the fluorescent protein gene mScarlet under the control of a tetracycline induced promoter. The mScarlet gene on each plasmid was tagged with a different protein degradation sequence of varying strength. Plasmid F contained the genetic circuitry for expression of the non-endogenous mf-Lon protease under the control of a lactose inducible promoter. Both plasmids also contained constitutive expression systems for their respective repressor proteins (TetR and LacI).
Upon receiving the DNA samples from William and Mary, plasmids A-E were cotransformed alongside plasmid F. Colonies from these plates were grown overnight to maximum culture density and then reinoculated into fresh LB broth at a 1:20 dilution. These cultures were grown at 37 C 220 rpm for about two hours to allow for the cells to reach log phase. Following this, each cotransformant culture was split in half with one half being induced with anhydrotetracycline (ATC) to a final concentration of 50 ng/mL while the other half was induced with ATC and IPTG to final concentrations of 50 ng/mL and 0.1 mM respectively. This meant that for each system (A-E + F) there were two testing groups. One where only mScarlet expression was induced and one where mScarlet was induced alongside the mf-Lon protease.
Following induction, samples were loaded into a 96 well plate and placed into an incubated plate reader set at 37 C. Fluorescence at an excitation of 569 nm with emission at 593 nm was measured every 10 minutes over a three hour time course. Shaking occurred between measurements. Fluorescence readings were then normalized against OD700 readings taken prior to and after the sampling period with the OD700 readings in the middle time points being approximated via linear regression.





Based on these graphs, we can see that steady states of fluorescence were achieved in several of the test groups after approximately 2 hours. These findings agree with the data collected by William and Mary, thereby acting to support the notion that their system is effective in controlling gene expression speed. It should be noted that the data collected for the B + F test does not appear to be reliable. Normalized fluorescence at time point 0 should have been identical between the two induction groups. The fact that it is not points to a high likelihood of experimental error in the testing of this particular device. Time was not available for the conduction of another test.
Testing Done by William and MaryIn return for the testing conducted by our team, William and Mary agreed to alter our copper biosensor part (BBa_K2477013) for use with their mf-Lon protease system. After sending our part to them, William and Mary created several variations on the biosensor by adding different protein degradation tags onto the end of the RFP gene. These protein degradation tags are termed A through F. By incorporating these modified parts into cells along with their mf-Lon system, they were able to show that gene expression speed could be altered. The results for the different tags they tested with our part can be seen in the graphs below.

Normalized Fluorescence of Protein Degradation Tagged Copper Biosensor Collected Using FACS. Time course assay of Copper Sensor pdt parts, values are steady state normalized geometric means of three biological replicates taken on the FL3 channel. Shaded region represents one geometric std above and below mean.

Fluorescence of Protein Degradation Tagged Copper Biosensor Collected Using Plate Reader. Functional plate reader assay of Copper Sensor pdt parts, values are geometric means of the fluorescence/OD600 of three biological replicates, excitation/emission 584/612.
The first graph above shows that, when induced with 1 mM copper, the modified copper biosensor constructs are capable of reaching steady protein levels at a faster rate than the original copper biosensor. While the increase in speed/stability is not drastic, it should be enough to improve the part in terms its original purpose. With the system being somewhat faster and more consistent, it will allow the part to be used in an educational setting where it can serve as a demonstration of synthetic biology's potential. For more on the potential uses of our copper biosensor, please refer to our metal detection page.
The second figure above consists of the plate reader data from a fluorescence time course of the modified copper biosensors induced with .5 mM copper and .01 mM IPTG to induce mf-Lon expression. Copper induction occurred about 240 minutes into the time course while IPTG induction occurred around 360 minutes into the time course. It can be seen that all variants reached steady state approximately 120-150 minutes after this. The final fluorescence levels vary across the different constructs showing the versatility of the system.
Collaboration ConclusionsOverall, the collaboration between the UMaryland and William and Mary iGEM teams has been very successful. We were able to help characterize their project while they were able to improve the function of one of our parts. This mutually beneficial relationship helped to further the efforts of both projects and served to highlight the importance of collaboration within iGEM and across science in general.
For further reading on the collaboration between UMaryland and William and Mary, take a look at William and Mary's collaboration page.
Mid-Atlantic Meetup
University of Virginia, 29 July 2017

By attending the the 2017 Mid Atlantic Meet-up hosted by the University of Virginia, the UMaryland iGEM team both discovered further improvements on all three projects and established collaboration opportunities with other teams. The attendees of the event included: William & Mary, UDelaware, UNC-Asheville, and Georgia State. Following the presentations, the UMaryland team was inspired by William & Mary’s human practices and outreach efforts in education.
In addition to testing each team’s plasmids, our teams discussed collaboration opportunities on curriculum planning and methods in improving our “Lab-in-a-box” design and protocol. We had the pleasure of talking to all teams to reconsider the implementation of the banana project and revising the metal detection project with a narrower scope for education purposes.

The Mid Atlantic Meet-up’s informative breakout sessions featured several speakers and through these talks, the UMaryland team learned about the growth of open community labs and the beginnings of commercial implementations of synthetic biology. We also met Elliot Roth, who introduced us to Dr. Robalino and his work in saving the Cavendish banana. The breakout sessions motivated us to develop a “Lab-in-a-box” focused on accessibility in small communities for students. From this meet-up, the UMaryland team gathered novel methods in both teaching synthetic biology and catering to our local communities.