Team:Tianjin/Design
/* OVERRIDE IGEM SETTINGS */
Design
Background
Human existence on earth is almost impossible without the heavy metals. Even though important to mankind, exposure to them during production, usage and their uncontrolled discharge in to the environment has caused lots of hazards to man, other organisms and the environment itself. Heavy metals can enter human tissues and organs via inhalation, diet, and manual handling. As the process of urbanization and industrialization goes deeper and deeper, heavy metal pollution, a noticeable threaten to almost all the creatures, has become an essential problem to solve.
According to our human practice, the situation of heavy metal pollution (copper and cadmium ions) is marked on a world map, and the severity of heavy metal pollution has been increasing all over this map. Places with serious pollution includes middle Asia, eastern Asia, southern Europe, and Latin America. In addition, not only fresh water sources, but also soil and crops are seriously contaminated by heavy metals. On average, during three out of ten suppers we have, we absorb excess heavy metals over the standard concentration.
Considering the rigorous situation we face, our team decided to design an advanced system for typical toxic heavy metal disposal based on Saccharomyces cerevisiae.
Mating-type Switch and Mating Switcher
Overview
After we found there might be revolutionary usage about the mating type switch (MTS) of yeasts in our heavy metal deposition system, In laboratory, the species of budding yeast we usually used are BY4741 and BY4742, whose HO gene are knocked out. Therefore, we intended to use two groups of MATa yeasts to realize the mating switcher.
One of these groups was required to achieve MTS. We decided to achieve MTS by introducing the HO gene into this group of yeasts— Saccharomyces cerevisiae (BY4741, in our lab, whose chromosome Ⅹhas been switched by synthetic chromosome Ⅹ. And it has been renamed as SynⅩsimilarly hereinafter.) . To make the MTS controllable, it is necessary for us to adopt inducible promoters to initiate the expression of HO gene or create a pathway functioning as single gene regulator. Eventually, we landed on the Gal1 promoter first, for its convenience and efficiency. As we read in R. Scott McIsaac’s work, their the rapid, tunable, single-gene specificity control system of single gene in yeasts has given us much impression. Therefore, we decided to use this system as one of our pathway designs for the expression of HO gene.
1. Getting the chassis.
Aiming to achieve MTS for environmental use, it is essential to make sure that when the MAT locus has DSB(double strands break) cleaved by HO, our type-a (MATa) yeast can only become type-α (MATα). Therefore, we used a Ura-tag to replace the HMR(a) domain in chromosome Ⅲ. In this way the HMR will no longer be the donor for the homologous recombination in the repairing process for MAT cleavage.
Since the change of mating type may appear successively, there is a great possibility that the same type haploid mate with each other. To avoid the existence of meaningless mating , we built an vector to express MATα genes to produce a1-α2 stable corepressor so that the haploid will regard itself as a diploid and prevent mating unless the MATa locus changes to the other one. After selection, by homologous recombination, we deleted the Ura-tag for further usage. We selected the target colonies(SynⅩdUra) via 5Foa plates.

Fig 1-1 Getting the chassis.
2. Construction of systems
(1) Gal System
In this pathway, we chose Gal1 as our promoter for the expression of HO gene, CYC1 as the terminator, and PRS416(with Ura-tag) as our vector. As for segments ligation, we design the cutting sites for Bsa1 enzyme in each part, hoping to achieve seamless ligation of these three parts.
We adopted the PCR method to amplify the Gal1-part and CYC1-part from a Gal1-Vika plasmid we had used in our former lab work. We designed specific primers for this procedure. After PCR, the Gal1 has the cutting sites for SalⅠand BsaⅠon both ends, and CYC1 has that for BsaⅠand BamhⅠon both ends. Meanwhile, the HO gene was obtained by gene synthesis, flanked by specific hangtags for BsaⅠto be cohesive with Gal1 upstream and CYC1 downstream. Thus, we have built our composite part (GHC).
After the ligation, we transformed the E.coli for the augment of the PRS416 plasmid with GHC. (GHC-416) And eventually, we transformed our SynⅩ-dUra for the GHC-416 to get our target yeasts——SynⅩ-dUra-416.
(2) Modified. Gal1 system
In this pathway, we introduced one kind of artificial transcription factor (ATF)—Z4EV into the regulation of HO gene expression. With Z4EV working with our Modified. Gal1 promoter, we hoped to reach the on off-target dynamic control of HO gene expression. Our designing for getting the Modified. Gal1-HO-CYC1 parts (MGHC) is quite the same as that for GHC mentioned above, only that we acquired our Modified. Gal1 part from the gene synthesis.

Fig 1-2 Modified. Gal1 system

As for the expression of Z4EV gene, we intended to induce it into the SynX chromosome in SynⅩ-dUra by homologous recombination. With overlap PCR strategy, we put an homologous domain CanA (originally from Can gene in chromosome X) in the upstream of the promoter of Z4EV. Thus, we got our CanA-TEF-Z4EV part. Then we planned to use Tdh2t as terminator attached with Leu-CanB (another part of Can gene).
Next, for double using the Leu-tag, we introduce Vika/vox system. We intended to attach the Vika operator (Tdh3p-Vikc-Tdh2t, TVT) following the Z4EV gene. We had two groups of yeasts, as mentioned above, one of them aimed to accomplish MTS and becoming MATα, the other with functional genes remained as MATa. According to our design, the former will express Vika recombinase, and the other contain functional genes whose expressions are controlled by vox-Terminator-vox structure. Thus, the function gene’s expression will be initiate during the cell fusion in yeast mating process.

Fig 1-3 Z4EV and Vika.
3. Test of MTS
In this section, we only got to test the Gal System due to time limits. And we figured that the results for Gal System is adequately enough to represent the feasibility of our designed strategy for MTS.
The whole test process can be divided into three steps.
Step 1:
Activate the Gal1 promoter. After that, the expression of HO gene in the SynⅩ-dUra-416 can be initiated.
Step 2:
Cultivate two groups of yeasts together. (one is SynⅩ-dUra-416, the other is normal BY4741 MATa) If the MTS has been accomplished (SynⅩ-dUra-416 can become MATα), the two groups of haploids can mate with each other and become diploids.
Step 3:
Test the results of mating by PCR method. We designed the primers for both MATa locus and MATα locus. The amplification of both MATa locus and MATα locus indicates that the yeasts has turned into diploids, the MTS has been achieved in other words.
Background
Mating switch of yeasts
Saccharomyces cerevisiae is a single-celled organism with three types, called a, α, and a/α. In Saccharomyces cerevisiae, three cell types differ from each other in their DNA content at the MAT locus which specifies the cell types. In nature, the two haploid cell types (a and α) of budding yeast are able to interconvert in a reversible manner by DNA-rearrangement with a DSB at the MAT locus, and this process is called mating-type switch.
The DSB at MAT locus is caused by HO endonuclease (a kind of site-specific endonuclease expressed by HO gene). DSBs in chromosomes can be repaired either by homologous recombination (HR) or by nonhomologous end-joining (NHEJ). In S.C haploids, the DSB caused by HO endonuclease mostly repaired by HR with HML(α) and HMR(a) as donors. If the donor is HML(α), the mating-type will become α, and vice versa. In this way, a haploid budding yeast is able to achieve mating-type switch.
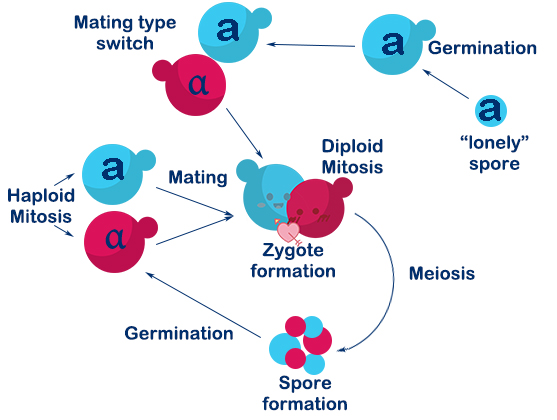
Fig 1-4 Mating switch of yeasts
Artificial Transcription Factors——Z4EV
Thanks to R. Scott McIsaac and Benjamin L. Oakes’s former work, we learned that Z4EV is a kind of fusion protein with three domains – DNA binding domain (DBD), estrogen receptor (ER) and VP16 activation domain. In the absence of β-estradiol, the ER interacts with Hsp90 chaperone complex and keep the ATF out of the nucleus. This AFT will provide a strong transcriptional activator that is dependent on the presence of β-estradiol. By using a synthetic 4-time-repeated zinc-finger DBD array from the mouse TF Zif268, residual off-target effects have been totally avoided.
Z4EV (the Z4EV gene has been induced into the SynX chromosome of this group of haploids) to strictly control the expression of HO gene. Unlike common β-estradiol-induced or galactose-induced promoters, this modified promoter is designed to be activated only when it is specifically bound with activated Z4EV factor.

Fig 1-5 Artificial Transcription Factors——Z4EV

Characterization of Mating Switcher (RFP to CRT)
Overview
Vika-vox system is used in our project in order to switch the expression from RFP to β-carotene, as a characterization of our Mating Switcher. In this way, we can easily visualize the function of our switcher through its color, as well as measure its efficiency and error rate.
Vika-vox system mainly consists of vox sites and reporting parts. At first the expression of RFP will be activating and the expression of β-carotene will be inhibited so that we can detect red fluorescence when recombinase vika doesn’t exist in Saccharomyces cerevisiae. After the expression of recombinase vika, with the deletion of RFP and terminators flanked by vox locus, β-carotene expresses and the strains take on an orange color. This is the whole characterization process of Mating Switcher.
Theoretical Background
1. Vika-vox system
Genome editing is emerging as a powerful technology platform which paved the way for exploring the nature of life comprehensively and systematically. Site-specific DNA recombinases have been tamed as a powerful tool in genome editing, such as Vika/vox and Cre/loxp. Site-specifc DNA recombinase Vika, originally identifed in a gram-negative bacterium Vibro coralliilyticus, could functionally and specifcally deleted genomic DNA fragment via recognizing specifc DNA site vox in yeast Saccharomyces cerevisiae and other spices, including mammal cell and bacteria.
Recently a milestone was reached with achieved total synthesis of functional chromosome Ⅴ DNA in yeast, Saccharomyces Cerevisiae. This chromosome owns lots of loxPxym sites. In spite of DNA sequence in recognizing site of loxP and vox shares a high similarity, it was demonstrated that activities of the two recombination systems were strictly independent to each other in yeast. Furthermore, the Vika/vox system functioned properly even in yeast cell carrying the synthesized chromosome on which lots of loxPsym sites are encoded.
2.Carotenogenic pathway
Carotenoids are a class of pigments of commercial interest that have important biological functions. Certain carotenoids can be synthesized by biotechnology, by either homologous or heterologous production. One example is that β-carotene can be overexpressed in S. cerevisiae., by introduced carotenogenic genes, crtE, crtI and crtYB, from the carotenoid-producing yeast Xanthophyllomyces dendrorhous. Like X. dendrorhous, S. cerevisiae is able to produce FPP and converts it into GGPP, the basic building block of carotenoids. Conversion of FPP into GGPP is catalyzed by GGPP synthase encoded by BTS1 in S. cerevisiae. Therefore, overexpression of only crtYB and crtI from X. dendrorhous in S. cerevisiae should generally be sufficient to transform S. cerevisiae into a β-carotene-producing organism. Additional overexpression of crtE from X. dendrorhous will increase GGPP levels and thereby enhance β-carotene production.

Fig.1. the carotenogenic pathway in X. dendrorhous

Experiment Design
1. Construction of vox-ura3-terminator-vox structure
We use synthetic chromosome Ⅴ of Saccharomyces cerevisiae to load our device, which is a single-celled organism called a. First of all, we use PCR to amplify basic parts including TEF promotor, ura3 gene, ura3-terminator and β-carotene gene. Among them, ura3 gene and ura3-terminator are flanked by vox locus. Then we use overlap PCR to combine these parts together. The next step is transform this composite part into Saccharomyces cerevisiae. We screen for the correctly transformed cell by using the Sc-Ura plate. For the purpose of verifying desired strain TVUVC, we use colony PCR to amplify the TEF promoter-vox-ura3 structure and ura3 terminator-vox-β-carotene structure. The length of the strip was observed by agarose gel electrophoresis.
2. Construction of vox-RFP-terminators-vox structure
This structure has a great similarity to the vox-ura3-terminator-vox structure above. Therefore, it is easy to construct because we only need to change the ura3 gene to the RFP gene. We use PCR to amplify five basic prats including TEF promotor, RFP gene, Adh1 terminator, ura3-terminator and β-carotene gene. Among them, RFP gene and ura3-terminator are flanked by vox locus. Then we use overlap PCR to combine these parts together. After that we use the lithium acetate conversion method to transfer this composite part into TVUVC. We screen for the correctly transformed cell by using the 5-FOA plate. This part will integrate into chromosomeⅤ by homologous recombination, and we will get another desired strain called TVRVC.
3.Verification of RFP in the TVRVC
The verification of RFP is carried out by using colony PCR to amplify the Homologous arm-TEF promoter-vox-RFP gene and terminators-vox-crtE gene, which determine the existence of vox sites and RFP gene. Then we can detect the red fluorescence.
4.Method of Red Fluorescence Assay
We used a variant of the mCherry red fluorescent protein (RFP). The variant sequence was codon-optimized for the expression in Saccharomyces cerevisiae as yeast-enhanced mRFP (yEmRFP) and can combine fluorescence and a purple visible phenotype. Unfortunately, the RFP can’t be directly observed by bare eyes, we decided to use the Fluorescence spectrophotometer and use OD600 to determination cell concentration. Meanwhile, we will observe using fluorescence microscopy for fluorescent proteins expression. The red color can be observed if yEmRFP is expressed.
5.Construction of vika System
We use a common expression vector plasmid, pRS416, to load vika part. First of all, we use corresponding restriction endonuclease Sal1 and Not1 to cut plasmid pRS416 and plasmid pRS415-vika, a gift from Y.J lab, and then use T4 DNA ligase to link them together, we can obtain the complete device we want. Finally, we transform this device into BY4742 by the lithium acetate conversion method, and we screen for the correctly transformed cell by using the Sc-Ura plate. BY4742 is a single-celled organism called α.
We also use another plasmid pRS413 by previous method.
6.The induction of Mating
The Saccharomyces cerevisiae called TVRVC is a single-celled organism called a. At first, we cultivate pRS416-vika in Sc-Ura medium without glucose for three hours. To induce the expression of vika, they will culture to saturation in Sc medium with raffinose and galactose for twelve hours. After that, recombinase vika are induced to express and we make α-pRS416-vika cell and a-TVRVC cell mate in YPD medium for eight hours. Two types cells are fused and form diploid yeasts, in which recombinase vika bind with vox locus, and then delete RFP gene and Adh1 terminator flanked by vox sites. After the Mating Switcher, β-carotene expresses and the color of cell will transform from white to orange. At last we smear yeast solution on Sc-Ura-Leu plate to screen for the correctly mating cell. We can judge the existence of recombinase vika by the color of the colony, and obtain the efficiency of mating.
7.Culture and Expression Condition of Saccharomyces cerevisiae in this experiment
Traditional YPD culture medium (22g/L glucose, 20g/L peptone, 10g/L yeast extracts) is used by us. Sc-Ura solid culture medium (22g/L glucose, 6.7g/L yeast nitrogen base, 1.224g/L nutrient deficiency mixture without Ura, His, Leu and Trp, 20g/L agar powder, 5mg/L Trp, His and Leu) is used to screen for correctly transformed cell. 5-FOA solid culture medium (22g/L glucose, 6.7g/L yeast nitrogen base, 1.224g/L dropout, 20g/L agar powder, 1ml/L His, Trip, Leu and 2.5ml/L Ura) is used to screen for correctly transformed cell. Sc medium with raffinose and galactose culture medium (20g/L raffinose, 6.7g/L yeast nitrogen base, 1.224g/L nutrient deficiency mixture without Ura, His, Leu and Trp, 20g/L agar powder, 10x galactose, 5mg/L Trp, His and Leu) is used to induce to express vika recombinase. Sc-Ura-Leu solid culture medium(22g/L glucose, 6.7g/L yeast nitrogen base, 1.224g/L nutrient deficiency mixture without Ura, His, Leu and Trp, 20g/L agar powder, 5mg/L Trp and His) is used to screen for the correctly mating cell. All the cells are cultured in 5mL medium at 30℃ with shaking speed of 220rpm.
Expected Results
In our design, Mating Switcher is a means of gene regulation. We can transform from one functional system to another system through this switch conveniently. To show the function of Mating Switcher more intuitively, we construct this RFP system to be a characterization.
First, we can successfully construct the corresponding expression vector.
Second, we expect to observe the expression of red fluorescent protein before switch is on. We expect red fluorescent protein to be observed under a fluorescence microscope. And it is desirable that the fluorescence spectrophotometer can measure a high and stable fluorescence value.
Third, vika-vox system can play a role on function conversion. It is means that β-carotene gene can express after mating. We will observe yellow colonies on nutrient label medium.
Reference
[1] Lin, Qiuhui et al. “Robust Orthogonal Recombination System for Versatile Genomic Elements Rearrangement in Yeast Saccharomyces Cerevisiae.” Scientific Reports 5 (2015).
[2] Karimova, Madina et al. “Vika/vox, a Novel Efficient and Specific Cre/loxP-like Site-Specific Recombination System.” Nucleic Acids Research 41.2 (2013).
[3] Verwaal, René et al. “High-Level Production of Beta-Carotene in Saccharomyces Cerevisiae by Successive Transformation with Carotenogenic Genes from X.anthophyllomyces Dendrorhous .” Applied and Environmental Microbiology 73.13 (2007): 4342–4350. PMC. Web. 27 Oct. 2017.
Resistance to Heavy Metals (SCRaMbLE)
Overview
Synthetic chromosome rearrangement and modification by loxP-mediated evolution(SCRaMble) generates combinatorial geomic diversity through rearrangements at designed recombinase sites. We applied SCRaMble to Saccharomyces cerevisiae(synX)to attain strains with better tolerance to high concentration of cadmium ion and cupric ion solution and compared the growing condition with the original strains to demonstrate the validity of SCRaMble.
Theoretical Background
Sc2.0 Project
Synthetic Yeast Genome Project (Sc2.0) is the world’s first synthetic eukaryotic genome project that aims to create a novel, rationalized version of the genome of the yeast species Saccharomyces cerevisiae. On March 10th, 7 articles related to Sc 2.0 were published on Science. As a member of Sc 2.0, YJ lab has completed two synthetic yeast chromosomes, and two articles are published on Science discussing about challenging but exciting task of building synthetic chromosomes V, X .
Cre-loxpsym System
As a part of the Sc2.0 Project , yeast chromosomes are targets, named loxPsym sites. loxPsym sites are substrates for Cre-EBD , which is an inducible form of the appropriate site-specific recombinase. Results of the recombination events are dependent on the directionality of the loxp sites. Because of the asymmetry of the loxp sites, when a pair of loxp sites is present in the same DNA strand and they are in opposite orientations, the Cre recombinase will catalyze the inversion of the gene in between the loxp sites; when the loxp sites are in same orientation, this gene will be deleted. Unlike the native directional loxP site, which permits a single orientation for recombination, the synthetic loxPsym site’s symmetry ensures that any pair of sites can recom- bine in either orientation. Then, controlled expression of Cre-EBD lead to deletions and inversions with chromosome segments flanked by loxPsym sites. This characteristic allows more possibilities of recombination on yeast chromosome which lives up to our expectations.
Experiment Design
Construction of Cre-loxpsym System
First, We use two common expression vector plasmid, PRS416 and PRS413(with different nutrition label), in Saccharomyces cerevisiae to load our device, which consists of heterologous gene part (PCLB2 promoter, Cre-EBD gene, CYC1 terminator) . Second, we transform the pSCW11-CRE/EBD plasmid into synX strain , respectively and get three strains with Cre-EBD, 079 and 160 with ura tag ,085 with his tag.
Then, under the cell culture environment with traces of estradiol(1μL of 5mM estradiol / 5mL media), three strains are incubated at 30℃for 6 hours. After dilute 1000 times and wash 2 times with water to remove estradiol and spot on on SC plates with gradient concentrations of copper ion and cadmium ion.
Finally, incubate plates for 3 days at 30℃ and observe the growth of strains.
Screening of Strains with High Tolerance
Aiming at screening strains with tolerance to high concentration of cadmium ion and cupric ion solution, we prepare SC culture medium with 3mM, 4mM, 4.5mM, 5mM, 5.5mM, 6mM, 6.5mM, 7mM copper ion and SC culture medium with 0.01mM, 0.05mM, 0.1mM, 0.15mM, 0.2mM cadmium ion. Though detecting the number, size of colony of different strains on SC plates with gradient concentrations. We further narrow the range of concentrations and screen strains with optimal characteristics.
Verification of Cre-EBD Effect (Dilution Assay and Measurement of cell survival rate)
To demonstrate the verification of Cre-EBD effect, we diluted optimal strains to 10-1、10-2、10-3、10-4、10-5 and made dilution assay on SC culture medium with copper ion and cadmium ion. Obviously if optimal strains grow better than other blanks, the answer is YES.
What's more, we measure the survivial rate of optimal strains with original strains synX. After cultivating yeasts in YPD overnight, we take 200μL culture medium into ultrahigh concentration copper ion and cadmium ion solution. Then, coate plate after 10min, 30min, 1h and 2h. Through counting the number of colony, we can obtain and compare the curve describing the cell survival rate of optimal strain and synX.
Detection and Enrichment of Copper ions (Parts’ Improvement)
Overview
This idea was inspired by the naturally-occurring metal-ion-induced promoters. Ligating this kind of promoters with a reporter gene such as RFP is a common idea to visibly monitor the concentration of metal ions.
In Saccharomyces cerevisiae S288C, there naturally exists a copper-induced promoter – CUP1p. The CUP1 promoter is activated by ACE1, a transcription factor which binds to copper ions. It is previously available as a standalone part as BBa_K945002, produced by Tec-Monterrery’s 2012 iGEM team, and team iGEM16_Washington modified this part with illegal restriction sites removed to make this part (BBa_K2165004) easier to control and operate. Our goal is to more perfectly characterize this promoter and improve it to create some handy parts.
Improvement
The characterization of this promoter wasn’t perfectly completed, so we first construct a biosensor based on CUP1 promoter (BBa_K2165004) and yEmRFP (BBa_K2407012) to improve the characterization and function of this BioBrick.
To improve this promoter, we first want to shorten this promoter with all core sequence retained. Compared with other promoters used in prokaryote, this promoter is much longer, which is not beneficial for BioBricks to easily assembly. Under this condition, we want to shorten its sequence as much as possible without functional changes.
In fact, this promoter is leaky in the absence of inducer. To make it more sensitive and lower the threshold of expression, we chose to transform the promoter with error-prone PCR. After finishing the library, the sensitivity and response rages of engineered promoters will be characterized by the fluorescence intensity.
After the detection, we will use Mating Switcher to rapidly open following gene’s expression. We can either overexpress CUP1 to enrich copper in the yeast cell or display the Metallothionein on the surface of budding yeast.
Separation of Different Ions (Copper and Cadmium ions)
OVERVIEW
The genetic circuit based on the Vika-Vox system in genetically-engineered yeast enables stepwise treatment of heavy metal ions by a switch from the expression of Cup1 metallothionein (copper accumulation) to the expression of LIMT metallothionein (cadmium accumulation). We measure the concentration of heavy metal ions in the supernatant at equal intervals to test the efficiency of our system.
BACKGROUND
Cup1, a kind of functional metallothionein, can combine with copper ions in yeast.
As a metallothionein in Littorina littorea, LIMT is capable of binding a large number of cadmium ions.
EXPERIMENT DESIGN
1) construction of S.C-Cu(Cd)
The TEF promoter, the Cup1 gene, and the Ura3 terminator are ligated together by overlap PCR, and then the sequence is integrated into vox-ura3-vox system by yeast's homologous recombination. 5-FOA plate helps us to screen the correct cell after transferring. Similarly, the LIMT gene and the Ura3 nutritional label are integrated into the same chromosome. Screened by SC-ura3, we complete genetic circuit as figure 5-1,5-2.showing below.

figure 5-1. the construction of expressing copper metallothionein.
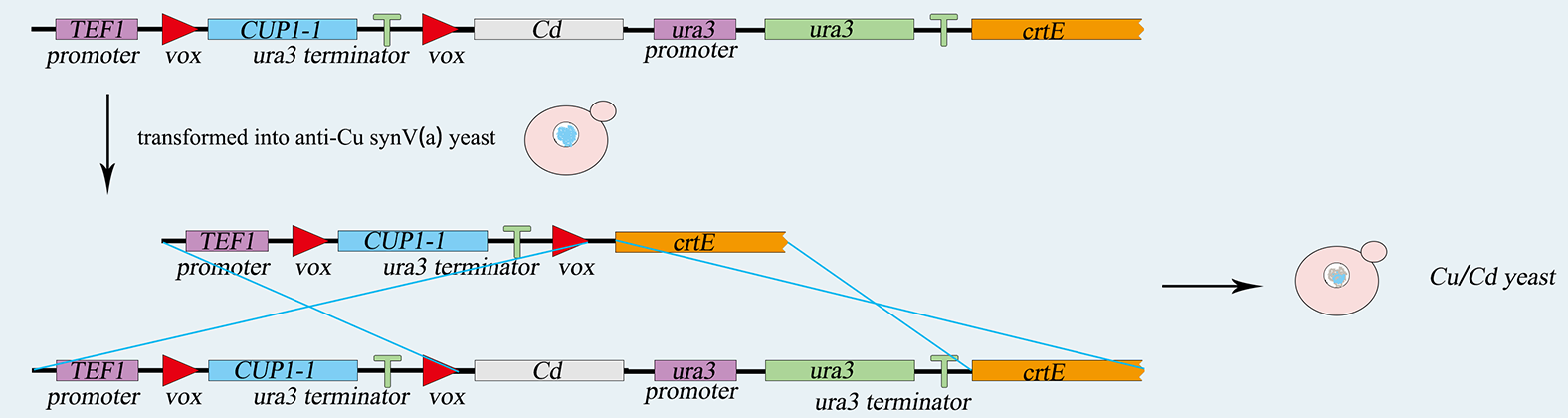
figure 5-2. the construction of expressing cadmium metallothionein.
2) Switch
A vector expressing the Vika recombinase is also transformed into the yeast strain. The expression of Vika enzymes can be induced by galactose. Then the Cup1 gene along with its terminator will be deleted by Vika recombinase hence the initiation of the expression of LIMT metallothionein. S.C-Cu is turned to S.C-Cd.
3) Copper Ions Accumulation
After completing the construction of the gene circuit, we start to test the efficiency of copper accumulation. Firstly, S.C-Cu, S8 (screened by SCRaMbLE) and BY4741 are all cultured in YPD liquid media for 24 hours for accumulation. after that the 430 mg/L copper ions solution is added to the media. The yeast cells are cultured in this solution for another 45 hours (30℃). Atomic absorption spectroscopy is used to measure the concentration of copper ions in the supernatant every 5 hours. According to the concentration change of copper ions at equal time intervals. We depict the adsorption curve of copper ions.
4) Cadmium Ions Accumulation
In order to reveal the yeast's influences on cadmium ions, we culture Cd Yeast, S1 and BY4741 in YPD liquid medium for 24 hours. After that the 16 mg/L cadmium ions solution is added to the media. The yeast cells are cultured in this solution for another 40 hours (30℃). The concentration of cadmium ions in the supernatant is measured every 5 hours.
5) Separation of Copper and Cadmium Ions
In the end, the accumulated S.C-Cu yeast strain is cultured in Sc medium with raffinose including copper ions and cadmium ions for 32 hours. Galactose is added after 12 hours of culture. Switch will be activated. The separation of Copper and Cadmium ions will be observed in heavy metal curve.
Reference
Wang J, Chen C. Biosorption of heavy metals by Saccharomyces cerevisiae: A review[J]. Biotechnology Advances, 2006, 24(5):427.
C. Baumann, A. Beil, S. Jurt, M. Niederwanger, O. Palacios, M. Capdevila, S. Atrian, R. Dallinger, O. Zerbe, Angew. Chem. Int. Ed. 2017, 56, 4617.
Dönmez G, Aksu Z. The effect of copper(II) ions on the growth and bioaccumulation properties of some yeasts[J]. Process Biochemistry, 1999, 35(1–2):135-142.