Difference between revisions of "Team:Newcastle/Results"
| Line 176: | Line 176: | ||
<h2 style="font-family: Rubik; text-align: left; margin-top: 1%"> Conclusions and Future Work </h2> | <h2 style="font-family: Rubik; text-align: left; margin-top: 1%"> Conclusions and Future Work </h2> | ||
| − | <p> | + | <p><i>E. coli</i> cells naturally have the C-P lyase pathway which degrades glyphosate into sarcosine. The fact that no formaldehyde was produced when glyphosate was added, but was when sarcosine was added, indicates that we have not overexpressed the C-P lyase pathway enough to produce enough sarcosine for sarcosine oxidase to convert into formaldehyde to be detected. |
</p> | </p> | ||
Revision as of 21:04, 27 October 2017
spacefill
spacefill
Our Experimental Results
Image will go here
Sarcosine Oxidase (Glyphosate to Formaldehyde)
BioBricks used: BBa_0123456 (New), BBa_7890123 (Team_Name 20XX)
Rationale and Aim
Sarcosine Oxidase (SOX) is an enzyme that oxidatively demethylates sarcosine to form glycine, hydrogen peroxide and formaldehyde (Figure 1) (Trickey et al. 1999). SOX was selected to be an example of a possible solution to one of the 5 problems in biosensor production that we identified - unconventional substrates. We defined an unconventional substrate as a substrate that we have little prior knowledge of but that can be adapted into something with an existing biosensor. SOX was specifically chosen to demonstrate that glyphosate, an unconventional substrate which there is not a lot information on, can be converted into formaldehyde which there are existing biosensors for (Ling and Heng 2010). As part of our project, SOX was designed to be an ‘adapter’ that could link glyphosate into our framework via a formaldehyde detector module. This concept could then be applied to other molecules that have easily detectable substrates in their degradation pathways. The aim of this part of the project was to demonstrate that SOX can be expressed by E. coli cells and that when glyphosate is added SOX can convert it to formaldehyde to be detected via a biosensor.

Figure 1: Biochemical pathway of the degradation of glyphosate to glycine and formaldehyde.
Background Information
Glyphosate is a herbicide that works by blocking the activity of the enzyme enolpyruvylshikimate-3-phosphate synthase (EPSPS), which converts carbohydrates derived from glycolysis and the pentose phosphate pathway to plant metabolites and aromatic amino acids. We attempted to design a system capable of glyphosate detection. With little information regarding mechanisms of glyphosate interactions with the cell, we could not design a simple system, representative of the majority of synthetic biology biosensor designs, in which a responsive transcription factor was able to affect the production of a reporter gene. The mining of transcriptome data has previously been used to find responsive DNA elements to a molecule of interest (Groningen 2012). Therefore, we analysed differences in transcriptome data between glyphosate sensitive and insensitive plants. A number of genes were found which were differently expressed. However, it was determined that it is more likely that this differential expression was not due to glyphosate directly, but rather the aromatic amino acid starvation caused by EPSPS inhibition by glyphosate, making these systems unsuitable for direct glyphosate detection. Various other systems we designed were also far from ideal, with high levels of complexity and reliance on native plant machinery. Through conversations with biosensor developers, we found that this problem was common in biosensor development - large amounts of often unavailable data is required for system design. For the Sensynova framework, we needed a more generic solution to this issue. Therefore, we expanded our search to look for biochemical reactions which we could monitor instead. This resulted in our concept of “adapter” devices which can alter difficult to sense molecules using biochemical reactions.
Design Stage
To ensure the codon usage of our SOX protein was not differing significantly from the average codon usage of E. coli, rare codons were removed from the sequence using the IDT codon optimisation tool to produce high protein expression. E. coli BL21-DE3 cells have higher levels of protein expression than DH5α cells and so were a more practical choice. This led to the expression of SOX being placed under the control of a T7 promoter due to BL21-DE3 cells producing T7 polymerase after the addition of IPTG. During the initial design stage of the protein, parts of the sequence were lost between optimisation and sending it to be synthesised into a gBlock. This was not discovered until expression of SOX was induced by IPTG in BL21-DE3 cells and a sample analysed by SDS-Page gel electrophoresis (Figure 2). It was noticed that the band we were expecting was of a lower weight than what it should have been; ~35kDa instead of ~42kDa. It was realised that the sequence in the PSB1C3 plasmid was different to the sequence origin. Therefore a new gBlock was synthesised using the proper sequence and an SDS-Page gel used to confirm that the protein expressed was of the correct weight (Figure 3).
Implementation
SOX was synthesised as a gBlock and assembled using HiFi Assembly. After assembly, SOX was transformed into E. coli DH5α cells and then into BL21-DE3 cells. This was done because DH5α cells are better for transformation, while BL21-DE3 cells are better for protein expression. Colonies indicated successful assembly, which was confirmed by creating plasmid DNA preparations of the colonies and performing confirmation digests to view on an agarose gel using the restriction enzymes Xba1 and Spe1 (Figure 2).

Figure 2: Restriction digest confirmation of first sarcosine oxidase assembly using Xba1 and Spe1 restriction enzymes.
The plasmid DNA preps with the correctly assembled SOX gBlock present were then transformed into E. coli BL21-DE3 cells. This was because BL21-DE3 cells are optimised for protein expression and because SOX was designed with a T7 promoter; DH5α cells do not produce the T7 polymerase required to express SOX whereas BL21-DE3 cells do in the presence of IPTG.
To prepare SOX for testing, cell cultures were grown following this protocolto step 4. Bradley’s CFPS protocol was then followed (link it). SDS-PAGE gel electrophoresis of the samples was done to check for SOX expression. 1 ml of each culture was lysed with lysozyme and incubated at room temperature before being boiled at 100° 10 minutes. 20 µl was loaded into each lane. At this point, an error was spotted with the size of SOX on the SDS-PAGE gel (Figure 3).

Figure 3: Lane 1: ladder, Lane 2: SOX, Lane 3: SOX+IPTG, Lane 4:SOX+IPTG, Lane 5:BL21 cells, Lane 6: sfGFP+IPTG, Lane 7: sfGFP
It was approximately 7 kDa too small. It was then discovered that the sequence synthesised as a gBlock was different to the original sequence found online; parts of the sequence were missing. A new gBlock with the correct sequence was synthesised and the above methods for assembly and preparation for testing were repeated (Figures 4 and 5).

Figure 4: Sarcosine Oxidase expression was induced by adding 40 µl of 100 mM IPTG. Lane 1: 6 µl ladder, Lane 2: 10 µl sfGFP, Lane 3: BL21-DE3, Lane 4: 10µl SOX 1, Lane 5: 10 µl SOX 2, Lane 6: 10 µl SOX 3, Lane 7: 10 µl SOX 4, Lane 8: 10 µl SOX 5, Lane 9: 10 µl SOX 6, Lane 10: 6 µl ladder.
To test for the presence of formaldehyde, and to demonstrate this part works, larger cultures were grown following the aforementioned protocols, and the cells harvested, washed and lysed by sonication. 0 µl, 20 µl, 200 µl and 2 ml of Glyphosate at 10 mg/L concentration was added to the cell lysate and incubated at 37°C. Every 2.5 hours the lysate was tested for the presence of formaldehyde with commercial formaldehyde testing strips.
After 8 hours of testing and left overnight, none of the samples had produced formaldehyde according to the testing strips. The testing strips detect a minimum formaldehyde concentration of 10 mg/L, so it was possible that formaldehyde had been produced but that there was too little of it to detect with the strips.
We decided to add Sarcosine instead of Glyphosate to determine whether the part was working. Everything was repeated the same but instead we added 0 µl, 50 µl and 200 µl of Sarcosine at 0.9 g/50 ml.
Characterisation
To determine whether sarcosine oxidase had been successfully expressed after adding IPTG we performed an SDS-Page gel. After inducing, harvesting and washing the cells 1 ml was taken from each culture to be loaded into the gel. The cells were lysed using lysozyme and boiled for 3 minutes at 100°C loading 10 µl into the gel (Figure 4) or lysed using lysozyme and boiled for 10 minutes at 100°C loading 20 µl into the gel (Figure 3). In both SDS-Page gels of the incorrect and correct SOX sequences (Figures 3 and 4 respectively) a band is present in the lanes that have been loaded with SOX induced with IPTG. Figure 6 shows that SOX works as expected, producing formaldehyde, however formaldehyde is produced even when SOX expression has not been induced with IPTG, indicating leaky expression.
Conclusions and Future Work
E. coli cells naturally have the C-P lyase pathway which degrades glyphosate into sarcosine. The fact that no formaldehyde was produced when glyphosate was added, but was when sarcosine was added, indicates that we have not overexpressed the C-P lyase pathway enough to produce enough sarcosine for sarcosine oxidase to convert into formaldehyde to be detected.
References
Ling YP, Heng LY (2010). A Potentiometric Formaldehyde Biosensor Based on Immobilization of Alcohol Oxidase on Acryloxysuccinimide-modified Acrylic Microspheres. Sensors 10:9963-9981. Trickey P, Wagner MA, Jorns MS, Mathews FS (1999). Monomeric sarcosine oxidase: structure of a covalently flavinylated amine oxidizing enzyme. Structure 7:331-345. Turgay U, Bakar M, Shearman RC, Budak H (2010). Genome-wide profiling and analysis of Festuca arundinacea miRNAs and transcriptomes in response to foliar glyphosate application. Molecular Genetics and Genomics 283:397-413. Zhu J, Patzoldt WL, Shealy RT, Vodkin LO, Clough SJ, Tranel PJ (2008). Transcriptome response to glyphosate in sensitive and resistant soybean. Agricultural and Food Chemistry 13:6355-63.
Synthetic Promoter Library
BioBricks used: BBa_0123456 (New), BBa_7890123 (Team_Name 20XX)
Rationale and Aim
The Sensynova multicellular biosensor platform has been developed to overcome the limitations identified by our team [hyperlink to human practices] that hamper the success in biosensors development. One of these limits regards the lack of modularity and reusability of the various components. Our platform design, based on the expression of three main modules (Detector, Processor and Output) by three E.coli strains in co-culture, allows the switch of possible variances for each module and the production of multiple customised biosensors. This section of the project is based on testing the modularity of the system by replacing the IPTG sensing unit present in the Sensynova platform with various synthetic promoters that are regulated by small molecules.
Background Information
Text goes here.
Design Stage
Text goes here.
Implementation
Text goes here.
Characterisation
Text goes here.
Conclusions and Future Work
Due to time constraints, we lacked the time to characterise these parts into the Sensynova platform within the lab.
References
Text goes here.
Arsenic Biosensor
BioBricks used: BBa_0123456 (New), BBa_7890123 (Team_Name 20XX)
Rationale and Aim
The Sensynova multicellular biosensor platform has been developed to overcome the limitations identified by our team [hyperlink to human practices] that hamper the success in biosensors development. One of these limits regards the lack of modularity and reusability of the various components. Our platform design, based on the expression of three main modules (Detector, Processor and Output) by three E. coli strains in co-culture, allows the switch of possible variances for each module and the production of multiple customised biosensors. This section of the project is based on testing the modularity of the system by replacing the IPTG detector part of the Sensynova design with different detecting parts. In particular, an Arsenic sensing part will be used.
Background Information
The part BBa_J33201 was made by the Edinburgh team in 2006.

Figure 1: Such and such
This part consists of the promoter of the E. coli JM109 chromosomal arsenic detoxification operon (ars operon), including the ArsR repressor binding site and the arsR gene encoding the arsR repressor protein, together with its ribosome binding site. Addition of any other genes to the 3' end of this part will result in their expression being dependent on the presence of sodium arsenate or sodium arsenite. Arsenite or arsenite anion binds to the repressor protein ArsR, resulting in inability to repress the promoter. Based on our experiments, a concentration of 1 micromolar sodium arsenate in LB is sufficient for essentially full expression, though this will vary according to conditions.
Design Stage
 Figure 2:
Such and such
Figure 2:
Such and such
 Figure 3:
Such and such
Figure 3:
Such and such
In order to introduce the Arsenic sensing part in the Sensinova framework, the part BBa_K2205008 containing the RBS B0034, the lasI coding sequence and the double terminator B0015 has been included in the design. The new part BBa_K2205022 presents biobrickable suffix and prefix and has been designed to have specific overhangs to be assembled in the plasmid pSB1C3 by Gibson assembly method. The part has been obtained by gBlock synthesis from IDT and subsequently assembled into the plasmid using NEB HI-Fi kit. The assembly mix was heat-shock transformed in competent DH5α and plated on Chloramphenicol LB plates. The colonies were tested through colony PCR and confirmed by sequencing.

Figure 4: Such and such
In the presence of arsenic, the repression will be avoided by binding the repressor ArsR This bound allows the transcription of the downstream gene, lasI. This gene encodes for the quorum sensing molecule C12, which acts as a connector to the processing cell.
Implementation
Text goes here.
Characterisation
Conclusions and Future Work
References
Brenner, K., Karing, D., Weiss, R. & Arnold, F. (2007) Engineered bidirectional communication mediates a consensus in a microbial biofilm consortium Proc Natl Acad Sci U S A 104(44): 17300 - 17304 de Mora K, Joshi N, Balint BL, Ward FB, Elfick A, French CE. A pH-based biosensor for detection of arsenic in drinking water. Anal Bioanal Chem. 2011 May; 400(4):1031-9. Epub 2011 Mar 27.
Psicose Biosensor (Evry Paris-Saclay Collaboration)
BioBricks used: BBa_K2205023 (New), BBa_??? (Evry Paris-Saclay 2017)
Rationale and Aim
The Sensynova multicellular biosensor platform has been developed to overcome the limitations identified by our team [hyperlink to human practices] that hamper the success in biosensors development. One of these limits regards the lack of modularity and reusability of the various components. Our platform design, based on the expression of three main modules (Detector, Processor and Output) by three E.coli strains in co-culture, allows the switch of possible variances for each module and the production of multiple customised biosensors. This section of the project is based on testing the modularity of the system by implementing the biosensor created by the 2017 Evry Paris-Saclay iGEM team into the Sensynova platform as part of our collaboration requirement.
Background Information
This biosensor was designed, made and submitted to the iGEM registry by the Evry Paris-Saclay 2017 team. We chose to use this system as a variant to the IPTG detector module present in the Sensynova platform in order to fulfil the requirement of collaborating with another iGEM team. The image below, provided to us by the Evry Paris-Saclay 2017 team, details the psicose biosensor design. It features the pLac derivative promoter pTAC (BBa_K180000), a RBS (BBa_B0034), the PsiR coding sequence, the terminator (BBa_B0015), the synthetic promoter pPsitac, a RBS (BBa_B0034), a mCherry coding sequence and finally the terminator (BBa_B0015) flanked by the iGEM prefix and suffix.

Figure 1: Such and such
The inducible system works as detailed in the diagram below. When pTAC is induced due to the presence of IPTG, PsiR is transcribed and binds to the pPsitac promoter repressing the transcription of the mCherry protein. When psicose is present, the sugar binds to PsiR, freeing up the promoter and subsequently the colour output

Figure 2: Such and such
Design Stage
In order to implement the psicose biosensor variant to the Sensynova platform, a design was created by replacing the IPTG sensing system in the original detector module with the construct detailed above, creating part K2205023. We chose to replace the pTAC promoter with the constitutive promoter present within the platform in order to eliminate the need for induction with IPTG. In place of the colour output present in the Evry Paris-Saclay design, we have added our part K2205008, which produces our first connector in order to trigger a response from following modules of the Sensynova platform.

Figure 3: Such and such
Part K2205023 detailed above was designed using Benchling and ordered for synthesis through IDT. Using Benchling, virtual digestions and ligations were simulated resulting in the plasmid map detailed below.

Figure 4: Such and such
Implementation
The Psicose detector construct obtained by gBlock synthesis has been designed to include required overhangs for Gibson assembly into the linearized plasmid pSB1C3. The plasmid backbone was acquired by digestion [Protocol link] of the part K2205015 with XbaI and SpeI, cutting out the original sfGFP construct. The Psicose detector construct was assembled into the plasmid backbone using the NEB Hi-Fi kit [Protocol link] and transformed into DH5α E. coli cells [Protocol link]. Colonies picked from streaked plates and cultures were prepared for miniprepping [Protocol link]. DNA samples were then sent off for sequencing [Website link] to ensure that the constructs were correct.
Characterisation
Text goes here.
Conclusions and Future Work
Due to time constraints resulted from synthesis delays, we lacked the time to co-culture this part with the Sensynova platform's multiple modules in order for the creation of variants. The part K2205023, the Evry Pasir-Sclay's psicose biosensor system as the detecting unit of the platform, has been submitted to the iGEM registry for future work and characterisation by future teams.
References
Text goes here.
Fim Standby Switch
BioBricks made and used: BBa_K2205005 (New), BBa_K1632013 (2015 Tokyo Tech part), BBa_K1632007(2015 Tokyo Tech part)
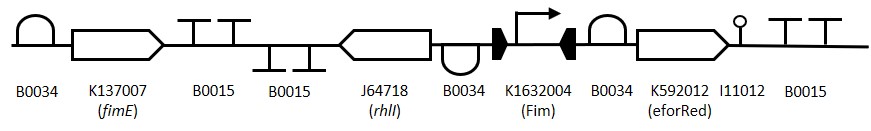
Figure 1: The Fim Switch in the native [OFF] state where the eforRED reporter is expressed allowing direct visualisation of the cells.
Rationale and Aim
Sensynova multicellular biosensor platform has been developed to overcome the limitations that hamper the success in biosensors development. One of these limits regards the lack of modularity and reusability of the various components. Our platform design, based on the expression of three main modules (Detector, Processor and Output) by three E.coli strains in co-culture, allows the switch of possible variances for each module and the production of multiple customised biosensors.
This part can be used within the platform as a Processor unit. Real world applications of biosensors are limited by many factors, one of which is that with most biosensors there is not a readout signal showing if the biosensor is working when not in use, i.e that the cells are still alive and have not lost their biosensor phenotypes. This can make them difficult to use, as well as market, since their viability comes into question as well as leading to false negatives/positives. Biosensors which rely on expression of a reporter signal may also suffer from unobserved activation due to weak or inconstant induction.
For this section of the project, as an improvement on a part by the Tokyo Tech team, we aim to produce a biobrick compatible part which is able to constitutively express a reporter signal prior to activation (to show that it is functioning) and to amplify a weak or inconsistent induction signal by permanently switching from an [OFF] to [ON] state after induction.
Background Information
Expression of the E. coli type 1 fimbriae gene is tightly regulated and phase dependent, i.e expression is either completely [ON] or [OFF] (Klemm., 1986). This change in expression is controlled by the action of two proteins FimB and FimE which independently act upon a 300bp promoter region upstream of the fimbriae gene. The 300bp promoter region is inverted to either activate or suppress expression (McClain et al., 1991). Typical gene regulation mechanisms rely on up or down regulation of a promoter from a baseline expression, the fimbriae mechanism of ‘ALL’ or ‘NONE’ makes it a useful tool for synthetic biology applications. While the FimB protein inverts the promoter back and forth between [ON] and [OFF] states the FimE protein permanently inverts the promoter from [ON] to [OFF]. This inversion can be used to amplify weak or inconsistent induction signals.
Since the part we are making is designed to amplify a weak signal which can then be detected by a downstream ‘reporter’ cell the quorum sensing system from P. aeruginosa was adapted to allow for signal transfer between cells. The rhlI gene from P. aeruginosa produces the quorum sensing molecule N-butyryl-AHL (C4-AHL) (Parsek et al.,2000), this molecule is membrane permeable and able to induce expression of a promoter upstream of sfGFP in another cell.
Fim 96 Plate assay Protocol
 Figure 2:
Representation of the switching mechanism of the Fim Switch, in the native [OFF] state the eforRED reporter is expressed (shown in red) allowing direct visualisation of the cells. Following the inversion of the promoter region (K1632004), eforRED expression is halted and the rhlI gene is expressed (J64718), this is now the [ON] state.
Figure 2:
Representation of the switching mechanism of the Fim Switch, in the native [OFF] state the eforRED reporter is expressed (shown in red) allowing direct visualisation of the cells. Following the inversion of the promoter region (K1632004), eforRED expression is halted and the rhlI gene is expressed (J64718), this is now the [ON] state.
Design Stage
To construct the Fim reporter switch 3 separate gBlocks were designed with overlapping adapter regions homologous to the iGEM prefix and suffix to allow for Gibson assembly into the pSB1C3 backbone whilst retaining biobrick compatibility. The individual genes and other components are shown in (Table 1). The 1st gBlock sequence starts with a RBS (B0034) upstream of the fimE ORF (K137007) with no promoter region, this is to allow for other promoters to be cloned in upstream of the part. Downstream of the fimE gene is a double terminator (B0015). All RBS and terminator sequences used are B0034 and B0015 respectively. The switching mechanism consists of the Fim promoter sequence (K1632004) flanked by two RBS-ORF-Terminator sequences. While in the native [OFF] state the Fim promoter drives expression of eforRed (K592012) and when flipped to the [ON] state drives expression of rhlI (J64718). The rationale behind using the fimE gene instead of is that it permanently inverts the promoter region meaning weak induction signals can be amplified by the Fim switch.
 Table 1:
Table 1:
Implementation
To assemble the Fim switch part the isothermal Gibson assembly cloning method was chosen as it would significantly shorten the time taken to assemble 3 separate sequences compared to traditional cloning methods. The 3 gBlock DNA fragments shown in (Table 1) were amplified by high fidelity Q5 PCR, the pSB1C3 backbone was digested with restriction enzymes EcoRI and PstI.
 Figure 3:
High fidelity amplification of the 3 gBlock fragments for assembly of the Fim Switch. The gBlock-1 amplification is shown in lanes 1+2 (819 bp), gBlock-2 amplification is shown in lanes 3+4 (1148 bp) and the gBlock-3 amplification is shown in lanes 5+6 (939bp).
Figure 3:
High fidelity amplification of the 3 gBlock fragments for assembly of the Fim Switch. The gBlock-1 amplification is shown in lanes 1+2 (819 bp), gBlock-2 amplification is shown in lanes 3+4 (1148 bp) and the gBlock-3 amplification is shown in lanes 5+6 (939bp).
The Gibson assembly reaction re-forms the iGEM prefix and suffix regions at the 5’ and 3’ ends of the Fim switch part making the component biobrick compatible while leaving no scarring regions. Following assembly, the plasmid was transformed into chemically competent DH5a E. coli https://static.igem.org/mediawiki/parts/b/b1/--T--Newcastle--MP--Protocol--Fim--96.pdf and colonies patched onto LB Chloramphenicol agar plates. A single patch showed the correct red colour indicative of the eforRed chromoprotein Figure 3.
 Figure 4:
Patches of the Fim Switch transformants. Patch number 6 shows the correct red colour which indicates expression of the eforRed chromoprotein.
Figure 4:
Patches of the Fim Switch transformants. Patch number 6 shows the correct red colour which indicates expression of the eforRed chromoprotein.
The red patch was cultured in LB chloramphenicol overnight and the plasmid DNA extracted by miniprep >>Protocol link<<. The plasmid was digested with restriction enzymes XbaI and PstI. The image in >>FigureX<< shows the DNA bands from the digested Fim switch plasmid.

Figure 5:
Restriction digestion of the Fim switch plasmid to confirm successful integration into the iGEM pSB1C3 backbone. The Fim switch plasmid (Lane 1) was digested with XbaI and PstI with expected band sizes of (2840 bp and 2044 bp). The pSB1C3 plasmid (Lane 2) containing sfGFP as a control was also digested with XbaI and PstI with expected band sizes of (811 bp and 2044 bp).
The Fim switch insert is 2882 bp in length which makes performing standard short sequencing reads challenging as multiple reactions are required to completely sequence the entire part. To overcome this we used our in-house Illumina MiSEQ to completely sequence the entire plasmid. Following quality control analysis the sequence was assembled and shown to be a match to the expected Fim switch part.
A problem we found with the Fim switch was that a subset of the colonies were prematurely switching from red to white. This is likely due to a low level of leaky expression of the fimE gene which then inverts the promoter region upstream of the eforRed gene. A single white colony was picked and cultured for use in downstream testing as a control as the switching of the promoter should express the rhlI gene and therefor produce the C4 quorum sensing molecule.
Characterisation
The fim Standby Switch has two main functions; a visual signal to show that the target compound has been detected and AHL production so that the part can be detected by a reporter cell. To characterise the part these functions are individually tested, in aim to further isolate issues if they occur.
To test the detecting function of the Fim Standby Switch it was assembled with the PBAD/AraC promotor. The PBAD/AraC promotor with Standby Switch parts were plated out onto four different LB plates containing chloramphenicol, two plates with different concentrations of glucose and chloramphenicol, and another containing arabinose. As colonies for the Fim Switch section were red and white when plated onto chloramphenicol plates due to leakiness, the plates with glucose in theory should suppress this switching and a greater percentage colonies on these plates should be red after transformation. The colonies on the arabinose plate should be white as translation of fimE leads to the flipping fimS, and expression of rhli.
The number of colonies on the plate that are white and red confirm inversion, this will show the percentage of colonies in the [ON] and [OFF] states. DNA sequencing will show inversion of the switch. As fimE is unidirectional over time the colonies should all become white on the plate containing arabinose. The plate containing glucose should repress leakage and the medium is supplemented by some percentage glucose.
To test the functionality of the Fim switch part, to ensure that C4 AHL is produced, the strain was cultured with the reporter strain >>REPORTER??<< which produces GFP in response to the quorum sensing molecule C4. Due to a small sub-population of the Fim switch strain being white, a single white colony was picked and cultured separately form the main culture. This culture is then co-cultured with a successfully independently tested reporter cell.This reporter cell detects C4 production and as a result GFP produced. This strain was used as a reference as it should produce activate expression of GFP in the reporter. This also shows that the reversed sequences for rhli and B0034 are working as expected. Since the issue of premature inversion of the Fim promoter may cause a problem with the Fim switch.

Figure 6:Initial Test of Fim Switch Red and white fim switch strains were spotted onto a lawn of the reporter strain (K2205015).

Figure 7:
Expression of GFP in the reporter (K2205015) strain in co-culture with the Fim switch strains. The assay was performed using methods described in >>Protocol<<. The data shows the expression of GFP in the reporter strain over a standard growth curve. The FimW and FimR strains represent the white and red variants of the Fim switch strain respectively, these were co-cultured with the reporter strain in a 1:14 ratio. Each data point is the mean of 3 biological repeats. RFU stands for relative fluorescence units.
Conclusions and Future Work
The aim of the Fim switch part was to make a processor module which can be visually inspected for functionality. The Fim switch has been shown to expresses the eforRed chromoprotein under normal (uninduced) conditions which allows the user to both determine that the strain is alive and has maintained the Fim switch plasmid. Following induction, the Fim promoter flips direction and begins expressing RhlI which synthesises the C4-HSL quorum sensing molecule. This has been shown to successfully induce expression of sfGFP in the reporter strain K2205015.Despite several attempts we were unable to produce a Fim switch testing construct where fimE expression could be controlled using the E. coli arabinose inducible promoter. Though transformations did yield some colonies when trying to make this part, none were red in colour. This possibly indicates that the arabinose inducible promoter (even when grown on 0.5% glucose) is still too active. The design for this construct has been submitted (BBa_K2205006).
Future Work: Since there is some leaky expression of the fimE gene (even without a promoter) to fine tune the Fim switch the lac operator could be inserted upstream of the fimE RBS to repress unwanted expression. The part could then be used following the addition of IPTG. An alternative method would be to clone a transcriptional terminator upstream of the fimE RBS to prevent leaky expression from elsewhere in the plasmid.
References
P. Klemm, Two regulatory fim genes, fimB and fimE, control the phase variation of type 1 fimbriae in Escherichia coli. EMBO J 5, 1389-1393 (1986).
M. S. McClain, I. C. Blomfield, B. I. Eisenstein, Roles of fimB and fimE in site-specific DNA inversion associated with phase variation of type 1 fimbriae in Escherichia coli. J Bacteriol 173, 5308-5314 (1991).
M. R. Parsek, E. P. Greenberg, Acyl-homoserine lactone quorum sensing in gram-negative bacteria: a signaling mechanism involved in associations with higher organisms. Proc Natl Acad Sci U S A 97, 8789-8793 (2000).
Signal Tuners
BioBricks used: BBa_K2205024 (New),BBa_K2205025 (New), BBa_K274371 (Cambridge 2009), BBa_K274381 (Cambridge 2009)
Rationale and Aim
The Sensynova multicellular biosensor platform has been developed to overcome the limitations identified by our team [hyperlink to human practices] that hamper the success in biosensors development. One of these limits regards the lack of modularity and reusability of the various components. Our platform design, based on the expression of three main modules (Detector, Processor and Output) by three E.coli strains in co-culture, allows the switch of possible variances for each module and the production of multiple customised biosensors. This section of the project is based on testing the modularity of the system by inserting two different sensitivity tuner constructs between the processing units of the Sensynova platform; BBa_K274371 and BBa_K274381.
Background Information
Both selected sensitivity tuner constructs were made and submitted to the iGEM registry by the Cambridge 2009 team. They were chosen as variants to the empty processing module present in the Sensynova platform due to the fact that, although they have been included in the iGEM distribution kit since their submission in 2009, they have yet to be successfully implemented into a team’s system, as far as we are aware. The 2007 Cambridge iGEM team built 15 different constructs that amplified the PoPS output of the promoter pBad/AraC detailed by image below taken from the Cambridge 2009 team's wiki.

The 2009 Cambridge iGEM team then re-designed these constructs to be PoPS converters, as image below taken from their wiki details, and generated a set sensitivity tuners corresponding to Cambridge 2007’s amplifiers.

BBa_K274371
This part is made up of an RBS (BBa_B0034), an org activator coding sequence (BBa_I746350) from P2 phage, the double terminator BBa_B0015 (made up of BBa_B0010 and BBa_B0012) and the inducible promoter PO (BBa_I746361) from P2 phage.

BBa_ K274381
This part is made up of an RBS (BBa_B0034), a pag activator coding sequence (BBa_I746351) from PSP3 phage, the double terminator BBa_B0015 (made up of BBa_B0010 and BBa_B0012) and the inducible promoter PO (BBa_I746361) from P2 phage.

Design Stage
In order to implement these two sensitivity tuner variants into the Sensynova platform, designs were made by inserting the above parts between the two constructs forming the empty processor module of our framework.

Using Benchling, virtual digestions of the two sensitivity tuners and ligations to the part K2205010, the connector 1 receiver module, were carried out. These two new constructs were then virtual digested and ligated to the part K2205011, the connector 2 reporter module, resulting in the two plasmid maps detailed below; parts K2205024 and K2205025.


Implementation
The sensitivity tuners parts BBa_K274371 and BBa_K274381 were requested from the iGEM parts registry. Upon arrival, parts were transformed in DH5α E. coli cells [Protocol link]. Colonies were picked and cultures were prepared for miniprepping [Protocol link]. Minipreps were digested [Protocol link] with XbaI and PstI for BioBrick assembly [Protocol link]. The part K2205010 contained in pSB1C3, was digested [Protocol link] using SpeI and PstI to allow for the insertion of the processing variants directly after the Las controlled promoter (pLas) that would trigger transcription of sensitivity tuners in the presence of connector 1 of the Sensynova platform. Ligations were set up overnight [Protocol link] using NEB’s T4 ligase and transformed in DH5α E. coli cells [Protocol link]. Colony PCR [Protocol link] was performed to check ligations. Colonies picked for this protocol were streaked onto a LB-agar plate. Colonies picked from streaked plates and cultures were prepared for miniprepping [Protocol link]. Minipreps were digested [Protocol link] with SpeI and PstI to allow for the insertion of the part K2205011 directly after the PO promoter. The part K2205010 contained in pSB1C3, was digested [Protocol link] using XbaI and PstI for BioBrick assembly [Protocol link]. Ligations were set up overnight [Protocol link] using NEB’s T4 ligase and transformed in DH5α E. coli cells [Protocol link]. Colony PCR [Protocol link] was performed to check ligations. Colonies picked for this protocol were streaked onto a LB-agar plate. Colonies picked from streaked plates and cultures were prepared for miniprepping [Protocol link]. DNA samples were then sent off for sequencing [Website link] to ensure that the constructs were correct.
Characterisation
Text goes here
Conclusions and Future Work
Due to time constraints, we lacked the time to characterise these parts into the Sensynova platform within the lab. The parts K2205024 and K2205025, the parts BBa_K274371 and BBa_K274381 respectively as processing units of the platform, were been submitted to the iGEM registry for future work and characterisation by future teams.
References
Text goes here
deGFP
BioBricks used: BBa_0123456 (New), BBa_7890123 (Team_Name 20XX)
Rationale and Aim
deGFP is a variant of Green Fluorescent Protein (GFP). It was initially designed by Shin and Noireaux (2010) for expression in cell-free protein synthesis (CFPS) systems and is more efficiently translated than other variants (e.g. eGFP). Through talks with other biosensor developers (for example, Chris French), and after reviewing legislation regarding the use of synthetic biology outside of the lab environment, the importance of CFPS systems as a chassis was highlighted. Despite its importance, CFPS systems can still suffer from some issues such as lower total protein synthesis than whole cells. By standardising and characterising a GFP variant which has been modified to have enhanced expression in these systems, it is hoped that CFPS will become a more attractive option for researchers. The aims for this section of the project were: (i) to standardise deGFP as a BioBrick and submit it to the iGEM repository, (ii) to demonstrate expression of deGFP in commercially available cell-free systems, and (iii) to characterise expression of deGFP in E. coli cells.
Background Information
deGFP is a modified variant of eGFP developed by Shin and Noireaux which is more efficiently translated in CFPS systems. It was designed by truncating the N-terminal sequence and introducing silent mutations which removed internal ribosome binding like sequences. The C-terminal sequence is also truncated as this has been shown to not be necessary for maximal fluorescence (Li et al. 1997). By removing ribosome binding like sequences, Shin and Noireaux have reduced the amount of incorrect ribosome binding events and hence increased translation efficiency. The length of the protein also contributes to enhanced translation efficiency by reducing the time and resources required for this process to reach completion.
Design Stage
The deGFP sequence was taken from the Addgene database (Plasmid #40019). The sequence was found to have no illegal restriction sites (i.e. no EcoRI, XbaI, SpeI, or PstI sites). A strong, standard Anderson promoter (J23100) and RBS (B0034) was added before the deGFP sequence with biobrick scar sites between each part. A double terminator (B0015) was added after the deGFP sequence. The entire construct was flanked by 30 bp overhangs with the pSB1C3 plasmid, such that the construct could be Gibson assembled into a plasmid digested with XbaI and SpeI. Extra bases were added between the overhangs and the construct so that once the part was assembled into the plasmid, the XbaI and SpeI sites could be regenerated and the biobrick prefix and suffix restored. This construct (J23100-deGFP) with the overhangs was submitted to IDT for synthesis as a gBlock. [BENCHLING LINK]
Implementation
The J23100-deGFP construct described above was Gibson assembled into a pSB1C3 plasmid using the NEB Hi-Fi assembly kit. To do this, pSB1C3 was digested with XbaI and SpeI to create a linearised plasmid backbone [LINK TO DIGEST PROTOCOL]. The deGFP gBlock DNA was prepared according to the IDT protocol [LINK HERE TO PROTOCOL] and assembled into the linear plasmid backbone according to the NEB Hi-Fi Protocol [LINK]. The assembly mixture was then transformed into commercial DH5α cells and incubated on chloramphenicol plates overnight [PROTOCOL LINK]. Colonies which were green under UV light were picked and grown in 5 mL LB broth overnight [PROTOCOL] before undergoing plasmid extraction [PROTOCOL]. Successful insertion of the J23100-deGFP construct into pSB1C3 was confirmed through a restriction digest with EcoRI and PstI followed by gel electrophoresis [PROTOCOL]. Figure 1 shows that the insert was successfully inserted as the double digest resulted in two linear bands at ~2000 bp (linear plasmid) and ~680 bp (deGFP). The DNA samples were then sent for sequencing to ensure that the construct was correct [DOWNLOAD LINK].
Characterisation
The expression of deGFP was firstly tested in E. coli cells using an experimental procedure similar to that used in the Interlab study. Cells transformed with pSB1C3-J23100-deGFP were grown in 10 mL LB broth overnight and OD600 nm was measured. Culture was added to 3 separate falcon tubes and made up to 12 mL with LB with chloramphenicol such that the starting OD600 of the culture was approximately 0.02. This set-up was repeated with cells containing an identical plasmid and construct, except sfGFP was in place of deGFP. As a control, untransformed cells were also prepared identically except the LB did not contain chloramphenicol. Tubes with only LB and LB+chloramphenicol were also prepared as blanks. The cultures were shake-incubated at 37oC. 300 μL samples from each tube were taken at time points of 15 mins, 2 hours, 4 hours, and 6 hours and stored at 4oC until the end of the experiment. 100 μL of each sample were then added to a 96-well plate. Fluorescence (excitation 485 nm, emission 510 nm) and absorbance (OD600 nm) were measured using a BMG-Labtech fluostar optima plate reader. Fluorescence intensity and grow rates for all three cell types were calculated over time (Figure 2). It was found that while cells expressing sfGFP had a much higher fluorescence intensity than cells expressing deGFP, the growth rate for cells with deGFP was closer to that of untransformed cells. This suggests that in vivo, either deGFP has lower expression than sfGFP, or each molecule of deGFP emits less fluorescence. INSERT 1 GRAPH Expression of deGFP in a cell-free protein synthesis (CFPS) system was then tested. Commercial S30 cell free kits for circular DNA from Promega were used so that results could be easily compared between labs. Reactions were prepared as described in the manufacturer’s protocol. Briefly, a master mix for 10 reactions was prepared (200 μL S30 premix, 150 μL S30 extract, 25 μL amino acid mix minus leucine, 25 μL amino acid mix minus cysteine) and 40 μL was added to nine tubes. To three tubes 1.2 μg J23100-deGFP was added, and J23100-sfGFP plasmid DNA was added to a further three. All reactions were then made up to 50 μL with nuclease-free water and transferred to a 96-well plate. Reactions were incubated in a BMG Labtech Fluostar Optima plate-reader at 37oC with fluorescence readings (excitation 485 nm, emission 510 nm) every 15 mins. It was found that, as expected, expression of the J23100-deGFP construct showed fluorescence above background level (Figure 4). It was also found that expression of the J23100-sfGFP construct in a CFPS system results in higher levels of fluorescence than deGFP. Therefore, although deGFP may show improved expression in CFPS systems than other GFP variants, sfGFP still shows higher fluorescence levels. INSERT 1 GRAPH
Conclusions and Future Work
Text goes here.
References
Text goes here.
Chromoproteins
BioBricks used: BBa_K2205016 (New),BBa_K2205017 (New),BBa_K2205018 (New), BBa_K1033915 (Uppsala 2013), BBa_K1033925 (Uppsala 2013), BBa_K1033929 (Uppsala 2013)
Rationale and Aim
The Sensynova multicellular biosensor platform has been developed to overcome the limitations identified by our team [hyperlink to human practices] that hamper the success in biosensors development. One of these limits regards the lack of modularity and reusability of the various components. Our platform design, based on the expression of three main modules (Detector, Processor and Output) by three E.coli strains in co-culture, allows the switch of possible variances for each module and the production of multiple customised biosensors. This section of the project is based on testing the modularity of the system by replacing the sfGFP output part of the Sensynova platform design with three different output chromoprotein variants; BBa_K1033929 (aeBlue), BBa_K1033925 (spisPink) and BBa_K1033915 (amajLime).
Background Information
All three selected chromoproteins were made and submitted to the iGEM registry by the Uppsala 2013 team. They were chosen as variants to the sfGFP present in the Sensynova platform as they exhibit of strong colour readily observed in both LB cultures and in agar plates when expressed. All three proteins have significant sequence homologies with proteins in the GFP family.
BBa_K1033915 – amajLime
The amajLime protein is a yellow-green chromoprotein extracted from the coral Anemonia majano. It was first extracted and characterized by Matz et al. under the name amFP486 (UniProtKB/Swiss-Prot: Q9U6Y6.1 GI: 56749103 GenBank: AF168421.1) and codon optimized for E coli by Genscript. The protein has an absorption maximum at 458 nm giving it a yellow-green colour visible to the naked eye.

BBa_K1033925 – spisPink
The spisPink protein is a pink chromoprotein extracted from the coral Stylophora pistillata. It was first extracted and characterized by Alieva et al. under the name spisCP (GenBank: ABB17971.1) and codon optimized for E. coli by Genscript. The protein has an absorption maximum at 560 nm giving it a pink colour visible to the naked eye. The strong colour is readily observed in both LB or on agar plates after less than 24 hours of incubation.

BBa_K1033929 – aeBlue
The aeBlue protein is a blue chromoprotein extracted from the basal disk of a beadlet anemone Actinia equine. It was first extracted and characterized by Shkrob et al. 2005 under the name aeCP597 and codon optimised for E. coli by Bioneer Corp. The protein has an absorption maximum at 597nm and a deep blue colour visible to the naked eye. The protein aeBlue has significant sequence homologies with proteins in the GFP family. The coding sequence for this protein was originally submitted to the registry as BBa_K1033916 by the 2012 Uppsala iGEM team.

Design Stage
In order to implement these three chromoprotein variants into the Sensynova platform, designs were made by replacing the sfGFP in the original reporter module with the parts detailed above that were ordered from the iGEM parts registry.

Using Benchling, virtual digestions of the three chromoproteins and ligations to the part K2205013, the connector 2 receiver module detailed above, were carried out resulting in the three plasmid maps detailed below; parts K2205016, K2205017 and K220518.



Implementation
The chromoproteins aeBlue (BBa_K1033929), amajLime (BBa_K1033915) and spisPink (BBa_K1033925) parts were requested from the iGEM parts registry. Upon arrival, parts were transformed in DH5α E. coli cells [Protocol link]. Colonies were picked and overnight cultures were prepared for miniprepping [Protocol link]. Minipreps were digested [Protocol link] with XbaI and PstI for BioBrick assembly [Protocol link]. The part K2205013 contained in pSB1C3, was digested [Protocol link] using SpeI and PstI to allow for the insertion of the chromoproteins directly after the RhI controlled promoter (pRhI) that would trigger transcription of colour proteins in the presence of connector 2 of the Sensynova platform. Stared colonies picked from streaked plates and cultures were prepared for miniprepping [Protocol link]. DNA samples were then sent off for sequencing [Website link] to ensure that the constructs were correct.
Characterisation
Text goes here.
Conclusions and Future Work
Text goes here.
References
Alieva, N., Konzen, K., Field, S., Meleshkevitch, E., Hunt, M., Beltran-Ramirez, V., Miller, D., Wiedenmann, J., Salih, A. and Matz, M. (2008). Diversity and Evolution of Coral Fluorescent Proteins. PLoS ONE, 3(7), p.e2680. Matz, M., Fradkov, A., Labas, Y., Savitsky, A., Zaraisky, A., Markelov, M. and Lukyanov, S. (1999). Nature Biotechnology, 17(10), pp.969-973. Shkrob, M., Yanushevich, Y., Chudakov, D., Gurskaya, N., Labas, Y., Poponov, S., Mudrik, N., Lukyanov, S. and Lukyanov, K. (2005). Far-red fluorescent proteins evolved from a blue chromoprotein fromActinia equina. Biochemical Journal, 392(3), pp.649-654.