Team:Oxford/Design
Project Design
Introduction
It was important to us that our design was as uncomplicated and elegant as possible. To effectively diagnose Chagas disease, we needed a system that could be performed in a field setting. This eventually pushed us in the direction of a cell-free system, inspired partly by the work of Keith Pardee in diagnosing the Zika virus cell-free.
Synthetic biology presents many possible paths, and we chose to investigate two of them: one DNA-based, one protein-based. The DNA-based system uses the TetR repressor, and the protein-based system uses OmpA and SpyTag/SpyCatcher to tether proteins to the outer membrane.
Background
There are many issues associated with developing a diagnostic for a neglected tropical disease. The method has to be (1) effective, (2) affordable, and (3) viable to perform and store within existing infrastructure. Additionally it needs to be accepted by the local community as a reliable and safe method for them to use. Currently, diagnostics for congenital Chagas disease do not sufficiently meet these requirements.
Synthetic biology is a currently underutilised as an option for diagnostics. Many tests use enzyme linked immunoassays (ELISAs), which can be expensive and difficult to conduct in some environments. They also rely on having common molecular epitopes, which may not be present for some diseases. Synthetic biology allows us to detect in a different way, and also circumvent the use of antibodies in some cases. It can also lead to smaller, faster, and cheaper detection. We conducted a survey of the general public and found that a majority were in favour of the development of a diagnostic using synthetic biology.
Cruzipain as a biomarker
Most current tests for Chagas disease focus on detecting the whole parasite, T. cruzi, its DNA, or antibodies produced in response to its biomarkers. We looked at a traditionally difficult biomarker, cruzipain(ref), a specific cysteine protease. Most tests failed to use it because it has many isoforms and no stable epitope, however its substrate was always the same, and we decided this was ideal to use for our system. This is because it is secreted by the parasite, and as the parasite is present in relatively high concentrations during the acute phase of the disease, there are likely to be relatively high concentrations of cruzipain present. It is also highly specific to T. cruzi meaning that we are unlikely to get false positives from this aspect. By looking at the protease directly we also circumvent a traditional problem which is diagnosis of newborn babies, who do not have fully developed immune systems and can therefore not use immunological tests. This developed our focus on congenital diagnosis. We developed two systems to detect it - one is DNA-based, one is OMV-based.
Design-Build-Test cycle
We used the engineering cycle from Imperial 2011 to help with the design, build, and testing of our system. This cycle shows how interactions with modellers, consumers, and the public contribute to the development of a system, and integrate with the wet lab work. The most important part is it is cyclical in nature, and a design may go through many cycles before it is finished. This constant dialogue is key to creating a product that matches the needs of the consumer as well as being scientifically sound. It allows for iterative improvement of the device.
First DBT cycle
Originally we envisioned producing hirudin directly from our system. However, results from our modelling data (see here) suggested that this would not be fast enough to prevent blood clotting. Thus, we needed an amplification system to increase the speed of hirudin production, and we settled on doing this by releasing it from a sterically hindered system by cleavage by TEV protease. From there, we considered the options of how we could control the levels of TEV protease in the system.

DNA approach
To detect cruzipain, its proteolytic function should lead to a detectable output. This leads to two logical options - a positive output and a negative output. As the system is DNA-based, and we want to control the production of an mRNA that would then be translated, this suggests either using an activating or repressing transcription factor. Cleavage by the protease to allow an activator to work would be a positive output, and cleavage to disable a repressor would be a negative output. After research, we concluded that it would be more feasible to produce a negative output using proteolytic cleavage.
Therefore we decided to research repressor proteins which could potentially be inactivated by cleavage from cruzipain. After reading several options, we decided on the TetR repressor(ref) as it was shown to be a very strong repressor, and had an easily accessible linker region between its two domains which we could modify to have a cruzipain recognition sequence.


TetR acts as a dimer(ref), and each monomer has a DNA binding domain and a dimerisation domain(ref)., consequently the Tet promoter has two TetR binding sites, both needed for full repression of transcription(ref). We took the DNA sequence of TetR Class B from BBa_K106669 and located the linker region using PyMOL and PDB entry 3zqi. The linker region was long enough to modify into a protease recognition sequence.
Due to the dangers of working with a protein from a pathogen, we decided to use a common laboratory protease instead to model our system and provide a proof-of-concept. Therefore, the TetR part we have designed (C200) has a Tobacco Etch Virus (TEV) protease recognition site instead of a cruzipain recognition site. The TEV protease is well-characterised(ref) and can be expressed in simple bacterial systems, making it ideal for testing our sensor.


We also believe that this version of TetR provides more versatility for relieving repression of a gene or other DNA sequence, as an alternative to adding tetracycline.
As a control, we also created a part with an unmodified TetR repressor (C250), which would give no output when the TEV protease is added. This enables us to understand the background rate of transcription and consequently the rate of false positive diagnoses from the repressor being ‘leaky’. Both TetR parts were created with CFP as fusion proteins so we could visualise them in a fluorescence plate-reader.
In addition to the TetR parts, we also created auxiliary parts to test them. We have designed a non-self-cleavable TEV protease from BBa_K1319004 with a His tag and an mCherry, and a DNA part which contains a medium-strength tet operator from BBa_R0040 in front of an RBS and eYFP (from AddGene).
The combination of CFP, mCherry, and YFP was chosen because they have very limited overlap in their absorption and emission frequencies, meaning it was easier to confidently quantify them relative to one another.
Overall, our proof-of-concept system consists of three parts:
- C100 - TEV protease, with or without mCherry, expressed in pBAD33
- C200 - TetR with a TEV protease cleavage site, with or without CFP, expressed in pQE-60
(C250 - TetR without a TEV protease cleavage site, with or without CFP, expressed in pQE-60) - C300 - Tet promoter, followed by an RBS and the sequence for YFP, expressed in pQE-60
Experimental Plan to test designs

We add IPTG to the cells to induce expression in pQE-60 of the TetR repressor, which then binds to the Tet site upstream of YFP.
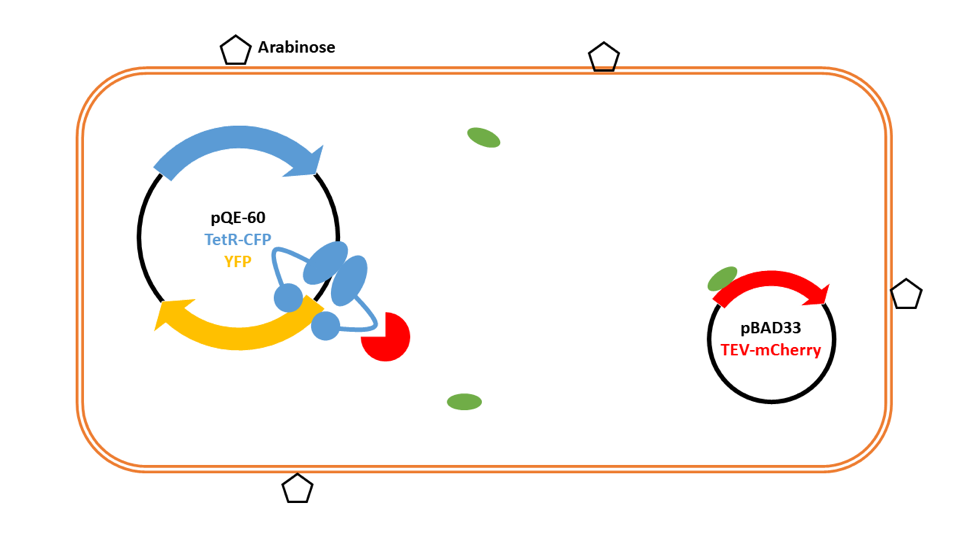
We remove IPTG and add arabinose to induce expression from pBAD33. This then produces a small amount of TEV protease in the system.

TEV protease then cleaves TetR at the linker.

TetR falls off the promoter region for the gene encoding YFP, allowing RNA polymerase to bind, leading to the production of YFP.

Lots of YFP is produced, and the small amount of TEV protease is amplified into a large signal. The fluorescence can be detected and quantified.
Experiments and data:
- C300 in expression vector to look at efficiency of the promoter
- Compare with C300 in shipping vector
- Measure YFP in C300 to find the time to detectable levels and its max expression
- Use C250 to see how much TetR we need for repression of C300
- Vary expression of C250 to find minimum TetR needed
- Only requires pQE-60 plasmid with C250 and C300
- Measure YFP and CFP with varied levels of inducer and time course in the plasmid with C200/250 and C300
- Compare C250 and C200 to check C200 is still functional
- Produce and purify TEV
- Experiments with two plasmids
- With optimal amount of C200/C250 induced, vary levels of C100 expressed
- Measure YFP, CFP and mCherry and compare YFP between C200 and C250
- Find levels of C100 that work to determine sensitivity in cells
- Vary time course to find optimal time for the system
- Compare above experiment to one with tetracycline instead of C100 vector to see how effective TEV protease relief of repression is
- Purify TetR
- Compare YFP levels for C200 and C250 in the above experiments with C200 and C250 with their fluorophores removed in the same experiments
Advantages and Disadvantages of the Cell-Free DNA System
| Advantage | Disadvantage |
|---|---|
| Amplification | Requires a large amount of cell machinery |
| No cells involved | Chance of DNA damage |
| Cheap | Chance of plasmid uptake by pathogens |
OMV System
Having identified some of the problems associated with the cell-free DNA- based system, we innovated a system that overcame these.
In our DNA-based system, TEV protease is activated in the presence of cruzipain; this mechanism relied, however, on DNA expression. We decided to generate an entirely protein-based circuit that acts in a similar way.
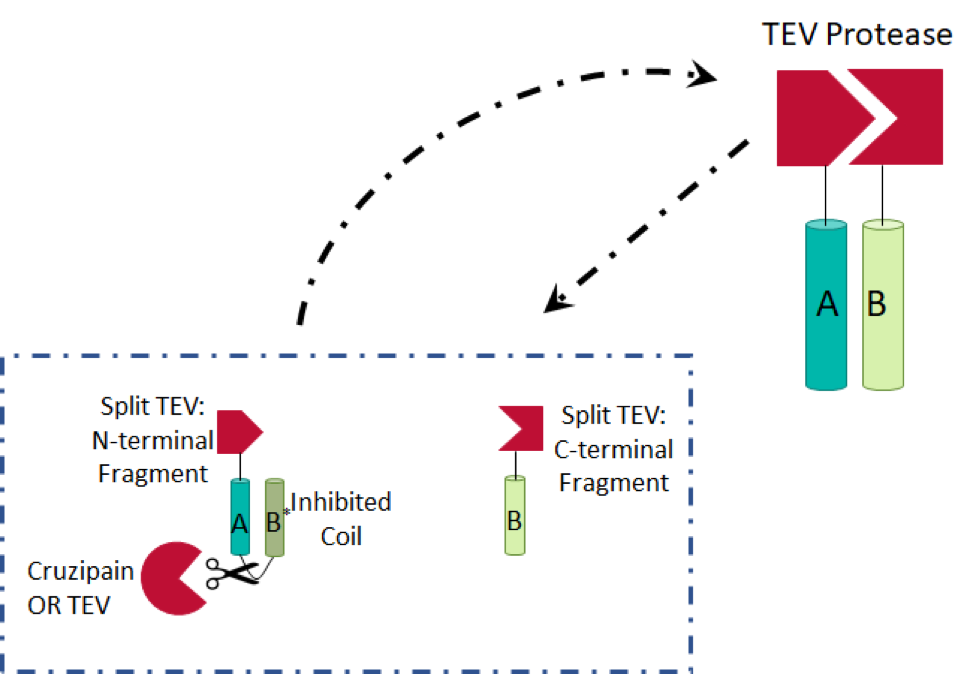
We initially considered sterically inhibiting TEV protease in the absence of cruzipain. However, due to the crystal structure of the protease, specifically the location of the N- and C-termini far from the active site, we decided this would not be a reliable method, with respect to false positives. We then discovered split TEV protease (Wehr et al.), in which two haves of the protein are present individually in an unconnected, and therefore inactive form. The halves are attached to dimerisation partners (with cruzipain/TEV protease cleavage sites in the linkers, in our device) to prevent spontaneous activation. In the presence of cruzipain, a protein circuit activating TEV protease molecules is induced: the TEV protease fragments are able to dimerize and amplify the signal. We combined these ideas with ones presented in a paper using split proteins in a protease detector (Shekhawat et al.) to generate the design, shown below. We generated two TEV protease fragments, each of which are bound to leucine zipper coils (A, B, and B*).

It is more entropically favorable for the B* coil to bind to the A coil, due to its position, and therefore dimerisation of the A and B coils is inhibited. Once the linker (A–B*) is cleaved, A binds to B, the enthalpically favored reaction. Two of the Leu residues in the B* leucine zipper have been mutated to Ala residues, decreasing the enthalpic benefit of binding to A (another Leu zipper) by approximately 0.2kJ/mol.
In this system, both our input (cruzipain) and our intermediate output (TEV protease) are proteases. We introduced an amplification step to improve the final output (active hirudin) of our circuit – the produced TEV protease will cleave A–B* linkers, thus increasing levels of TEV protease and inactive hirudin, to activate it. As a result, our system will require only a small initial signal to produce a clear response.
Due to the protein-based nature of our circuit, we would likely not be able to replicate it in a functional cell-free extract. We found a solution in outer membrane vesicles (OMVs). OMVs are lipid vesicles (made from the outer membrane) containing periplasmic solution and proteins constitutively made by all Gram-negative bacteria. A recent paper by Alves et al. demonstrated that enzymatic function could be protected during freeze-drying cycles by containing the enzyme in an OMV. We aimed to target the proteins in our circuit to the bacterial outer membrane, extract OMVs containing them, freeze-dry the OMVs for storage and transport, and finally re-solubilise and lyse the OMVs for diagnosis.
As the bacterial outer membrane transport system is still not yet fully understood, we adopted the technique described in Alves et al.: (1) OmpA, a membrane protein in E. coli which is known to be transported into OMVs at a high rate, is fused to SpyTag and (2) the functional circuitry component is fused to SpyCatcher and a TorA leader sequence. The TorA leader sequence will transport our protein into the periplasmic space (via the Tat translocase system), where SpyTag and SpyCatcher can form an isopeptide bond and fuse the two parts (1 and 2). The circuit proteins are then taken up into the OMV with OmpA. This system is shown in the diagram below.
Once in the OMVs, they can be freeze-dried, and then resolubilised when the diagnostic is needed. As our test was a blood test this would happen as soon as the blood was added. The OMVs then need to be lysed to expose the protein system within. Our proposal for this is to add powdered detergent to the protein system so that when it is solubilised the detergent can lyse the outer membrane vesicles. In order to test OMV lysis without lysing protein we designed the two parts shown below:

Biobrick: BBa_K2450401

Biobrick: BBa_K2450501
The OmpA protein is able to be inserted into the outer membrane as the SpyTag motif is very small. The quenched sfGFP can then bind to SpyTag and be taken up into OMVs as explained above. This results in the protein complex shown below:
