Team:Newcastle/Results
spacefill
spacefill
Our Experimental Results
Below is a diagram of our Sensynova Framework. Clicking on each part of the framework (e.g. detector modules) links to the relevant results.
Alternatively, at the bottom of this page are tabs which will show you results for every part of the project
Framework |
Framework Chassis |
|||
|
Biochemical Adaptor 
|
||||
|
Target 
|
|
Detector Modules 
|
Multicellular Framework Testing 
|
|
|
|
||||
|
C12 HSL: Connector 1 
|
||||
|
|
||||
|
Processor Modules 
|
||||
|
|
Framework in Cell Free Protein Synthesis Systems 
|
|||
|
C4 HSL: Connector 2 
|
||||
|
|
||||
|
Reporter Modules 
|
||||
|
Looking for Interlab Study related results? Click below! 
|
||||
Sarcosine Oxidase
BioBricks used: BBa_K2205003 (New), BBa_K2205004 (New)
Rationale and Aim
Sarcosine Oxidase (SOX) is an enzyme that oxidatively demethylates sarcosine to form glycine, hydrogen peroxide and formaldehyde (Figure 1) (Trickey et al. 1999). SOX was selected to be an example of a possible solution to one of the 5 problems in biosensor production that we identified - unconventional substrates. We defined an unconventional substrate as a substrate that we have little prior knowledge of but that can be adapted into something with an existing biosensor. SOX was specifically chosen to demonstrate that glyphosate, an unconventional substrate which there is not a lot information on, can be converted into formaldehyde which there are existing biosensors for (Ling and Heng 2010). As part of our project, SOX was designed to be an ‘adapter’ that could link glyphosate into our framework via a formaldehyde detector module. This concept could then be applied to other molecules that have easily detectable substrates in their degradation pathways. The aim of this part of the project was to demonstrate that SOX can be expressed by E. coli cells and that when glyphosate is added SOX can convert it to formaldehyde to be detected via a biosensor.

Background Information
Glyphosate is a herbicide that works by blocking the activity of the enzyme enolpyruvylshikimate-3-phosphate synthase (EPSPS), which converts carbohydrates derived from glycolysis and the pentose phosphate pathway to plant metabolites and aromatic amino acids. We attempted to design a system capable of glyphosate detection. With little information regarding mechanisms of glyphosate interactions with the cell, we could not design a simple system, representative of the majority of synthetic biology biosensor designs, in which a responsive transcription factor was able to affect the production of a reporter gene. The mining of transcriptome data has previously been used to find responsive DNA elements to a molecule of interest (Groningen 2012). Therefore, we analysed differences in transcriptome data between glyphosate sensitive and insensitive plants. A number of genes were found which were differently expressed. However, it was determined that it is more likely that this differential expression was not due to glyphosate directly, but rather the aromatic amino acid starvation caused by EPSPS inhibition by glyphosate, making these systems unsuitable for direct glyphosate detection. Various other systems we designed were also far from ideal, with high levels of complexity and reliance on native plant machinery. Through conversations with biosensor developers, we found that this problem was common in biosensor development - large amounts of often unavailable data is required for system design. For the Sensynova framework, we needed a more generic solution to this issue. Therefore, we expanded our search to look for biochemical reactions which we could monitor instead. This resulted in our concept of “adapter” devices which can alter difficult to sense molecules using biochemical reactions.
Design Stage
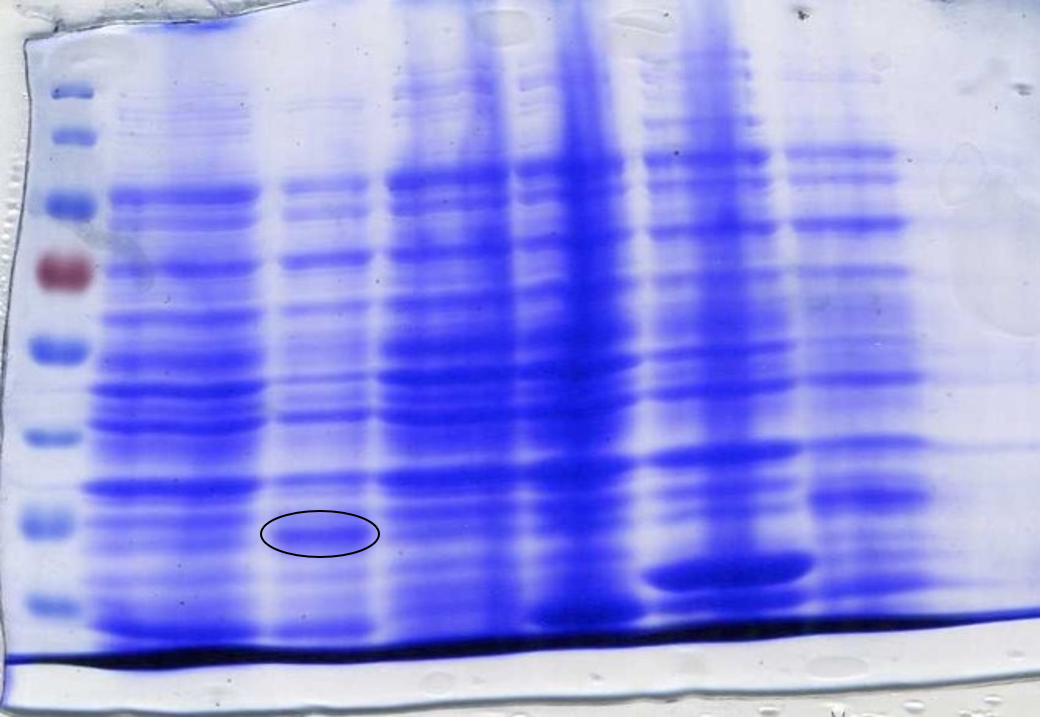
To ensure the codon usage of our SOX protein was not differing significantly from the average codon usage of E. coli, rare codons were removed from the sequence using the IDT codon optimisation toolto produce high protein expression. E. coli BL21-DE3 cells have higher levels of protein expression than DH5α cells and so were a more practical choice. This led to the expression of SOX being placed under the control of a T7 promoter due to BL21-DE3 cells producing T7 polymerase after the addition of IPTG. During the initial design stage of the protein, parts of the sequence were lost between optimisation and sending it to be synthesised into a gBlock. This was not discovered until expression of SOX was induced by IPTG in BL21-DE3 cells and a sample analysed by SDS-Page gel electrophoresis (Figure 2). It was noticed that the band we were expecting was of a lower molecular weight than what it should have been; ~35kDa instead of ~42kDa. It was realised that the sequence in the PSB1C3 plasmid was different to the sequence origin. Therefore a new gBlock was synthesised using the proper sequence and an SDS-Page gel used to confirm that the protein expressed was of the correct molecular weight (Figure 3).

Implementation
SOX was synthesised as a gBlock and assembled using HiFi Assembly. After assembly, SOX was transformed into E. coli DH5α cells and then into BL21-DE3 cells. This was done because DH5α cells are better for transformation, while BL21-DE3 cells are better for protein expression. Colonies indicated successful assembly, which was confirmed by creating plasmid DNA preparations of the colonies and performing confirmation digests to view on an agarose gel using the restriction enzymes Xba1 and Spe1 (Figure 4).

The plasmid DNA preps with the correctly assembled SOX gBlock present were then transformed into E. coli BL21-DE3 cells. This was because BL21-DE3 cells are optimised for protein expression and because SOX was designed with a T7 promoter; DH5α cells do not produce the T7 polymerase required to express SOX whereas BL21-DE3 cells do in the presence of IPTG.
To prepare SOX for testing, cell cultures were grown following this protocol to step 4. The protocol used for CFPS extract preparation [LINK IT] was then followed. SDS-PAGE gel electrophoresis of the samples was done to check for SOX expression. 1 ml of each culture was lysed with lysozyme and incubated at room temperature before being boiled at 100°C for 10 minutes. 20 µl samples were loaded into each lane. At this point, an error was spotted with the size of SOX on the SDS-PAGE gel (Figure 2).
The band was approximately 7 kDa too small. It was then discovered that the sequence synthesised as a gBlock was different to the original sequence found online; parts of the sequence were missing. A new gBlock with the correct sequence was synthesised and the above methods for assembly and preparation for testing were repeated (Figure 3).
To test for the presence of formaldehyde, and to demonstrate this part works, larger cultures were grown following the aforementioned protocols, and the cells harvested, washed and lysed by sonication. 0 µl, 20 µl, 200 µl and 2 ml of Glyphosate at 10 mg/L concentration was added to the cell lysate and incubated at 37°C. Every 2.5 hours the lysate was tested for the presence of formaldehyde with commercial formaldehyde testing strips.
After 8 hours of testing and left overnight, none of the samples had produced formaldehyde according to the testing strips. The testing strips detect a minimum formaldehyde concentration of 10 mg/L, so it was possible that formaldehyde had been produced but that there was too little of it to detect with the strips.
We decided to add Sarcosine instead of Glyphosate to determine whether the part was working. Everything was repeated the same but instead we added 0 µl, 50 µl and 200 µl of Sarcosine at 0.9 g/50 ml.

This shows SOX works as expected, however there is leaky expression as formaldehyde is produced when no IPTG is added.
Characterisation
To determine whether SOX had been successfully expressed after adding IPTG we performed an SDS-Page gel. After inducing, harvesting and washing the cells 1 ml was taken from each culture to be loaded into the gel. The cells were lysed using lysozyme and boiled for 3 minutes at 100°C loading 10 µl into the gel (Figure 3) or lysed using lysozyme and boiled for 10 minutes at 100°C loading 20 µl into the gel (Figure 2). In both SDS-Page gels of the incorrect and correct SOX sequences (Figures 2 and 3 respectively) a band is present in the lanes that have been loaded with SOX induced with IPTG. Figure 5 shows that SOX works as expected, producing formaldehyde, however formaldehyde is produced even when SOX expression has not been induced with IPTG, indicating leaky expression.
Conclusions and Future Work
E. coli cells naturally have the C-P lyase pathway which degrades glyphosate into sarcosine. The fact that no formaldehyde was produced when glyphosate was added, but was when sarcosine was added, indicates that we have not overexpressed the C-P lyase pathway enough to produce enough sarcosine for SOX to convert into formaldehyde to be detected.
References
Ling YP, Heng LY (2010). A Potentiometric Formaldehyde Biosensor Based on Immobilization of Alcohol Oxidase on Acryloxysuccinimide-modified Acrylic Microspheres. Sensors 10:9963-9981. Trickey P, Wagner MA, Jorns MS, Mathews FS (1999). Monomeric sarcosine oxidase: structure of a covalently flavinylated amine oxidizing enzyme. Structure 7:331-345.
Synthetic Promoter Library
BioBricks used: BBa_J61002 (Arkin Lab 2006)
Rationale and Aim
The Sensynova multicellular biosensor platform has been developed to overcome the limitations identified by our team [hyperlink to human practices] that hamper the success in biosensors development. One of these limits regards the lack of modularity and reusability of the various components. Our platform design, based on the expression of three main modules (Detector, Processor and Output) by three E.coli strains in co-culture, allows the switch of possible variances for each module and the production of multiple customised biosensors. This section of the project is based on testing the modularity of the system by replacing the IPTG sensing unit present in the Sensynova platform with various synthetic promoters that are regulated by small molecules.
Background Information
Promoter libraries can be created by varying many different as-pects of a wildtype promoter such as the upstream element prior to the -35 region, the downstream element, after the -10 region prior to -1, and its core sequence, between the -35 and -10 regions (Schlabach et al., 2010). In this study, we propose to use the PLac promoter sequence as our wildtype for creating promoter designs varying different areas of its sequence. One of such variation will be the substitution of the -35 and -10 currently found in PLac with the -35 (TTGACA) and -10 (TATAAT) regions found to be the most commonly occurring in E. coli natural promoters (Hawley and McClure, 1983, DeBoer, 1985, Harley and Reynolds, 1987). These were chosen to be the constant region between different promoter designs.

Figure 1: Graph Indicating the Most Frequent -35 and -10 Regions Found in E. coli Promoters. This image was taken from Harley and Reynolds (1987).
By analyzing the findings of Harley and Reynolds (1987) and Lisser and Margalit (1993), the decision to vary the number of base pairs in the region present between the -35 and -10 elements to 17 base pairs instead of the 18 present in the wildtype PLac. Variations of the upstream and downstream regions where the lac operon would normally bind to will also be investigated in this study by the production of three different promoter designs resulting in a diverse promoter library.

Figure 2: Graph Indicating the Most Frequent Spacer Between -35 and -10 Regions Found in E. coli Promoters. This image was taken from Harley and Reynolds (1987).
Design Stage
As seen in the image above (Image 3B), the regions known to be important for a reliable promoter expression (-35 and -10 regions) were changed to variant of the wildtype but kept constant between the three distinctive designs. These regions were discovered to be the most frequent occurring -35 and -10 regions in native E. coli promoters by Harley and Roberts in 1987. The sequences between such converged regions were kept constant as per the wildtype for designs 2 (P2) and 3 (P3). For design 1 (P1) however, they were randomized in order to test its effect. The decision to reduce the number of base pairs from 18, found in PLac, to 17 was made due to the results of the study by Harley and Roberts in 1987, listing this number to be the most frequent occurring number of base pairs gap found in regions in native E. coli promoters.
Design 1 (P1) was made by randomizing all elements of the promoter while only keeping the -35 and -10 regions constant. The upstream element (US element) of P2 were randomized while keeping the downstream element (DS element) conserved as per wildtype. The DS element of P3 however, was randomized while keeping the upstream element conserved. This systematic approach of randomization was chosen as it allows for the most variation between promote designs allowing for a rich synthetic promoter library.

Figure 3: Image Detailing Promoter Designs.
In order to implement these synthetic promoter detector variants into the Sensynova platform, designs were made by replacing the IPTG detection module of our framework with the promoter library.

Figure 4: SBOL Visual Detailing Detector Promoter Variants
Implementation
The promoter designs were sent off for synthesis by IDT as single stranded oligos.