|
|
| (24 intermediate revisions by 3 users not shown) |
| Line 1: |
Line 1: |
| | {{Aix-Marseille|title=EPS Depolymerase|toc=__TOC__}} | | {{Aix-Marseille|title=EPS Depolymerase|toc=__TOC__}} |
| | | | |
| − | To combat the problem that is ''Xylella fastidiosa'' we firstly started searching for natural solutions against this bacterium and surely, we found bacteriophages that loved destroying this bacterium.
| + | [[File:T--Aix-Marseille--EPSd.png|450px|right|thumb|EPS Depolymerase process to clean the xylem vessel from EPS.]] |
| | | | |
| − | In our researcher, a modified phage would not be possible because of restriction in spreading modified organism able to replicate in the wild. Therefore, a non-lytic and non-lysogenic phage was born using fibrous phage M-13 with a modification of its genome in the objective of being modular and flexible to use against varies pathogens. The phages against ''Xylella fastidiosa'' have a devious way to infect the bacterium by cleaving the sugar bond in the biofilm then link on the receptors pili type IV on the membrane that will sets off a series of action leading to the injection of the bacteriophage genome in the bacterium triggering the life cycle of the phage.
| + | To fight [[Team:Aix-Marseille/Xylella_fastidiosa|''Xylella fastidiosa'']] we searched for natural solutions. |
| − | A number of articles described the possibility of using these phages to eradicate ''Xylella fastidiosa'' but in my mind, that was an endeavor bigger than my capacities. One article talked about the EPS-Depolymerase as a potential tool for control of fire blight a disease caused by ''Erwinia amylovora''. | + | Some phages have a devious way to attack bacteria, digesting the biofilm so that they can their target more easily <ref>Vandenbergh, P. A., Wright, A. M. & Vidaver, A. K. Partial Purification and Characterization of a Polysaccharide Depolymerase Associated with Phage-Infected ''Erwinia amylovora''. Appl. Environ. Microbiol. 49, 994–996 (1985)</ref>. |
| | + | We decided to try to use this approach to attack [[Team:Aix-Marseille/Xylella_fastidiosa|''Xylella fastidiosa'']]. |
| | + | We found an enzyme called EPS-depolymerase <ref>Kim, W. S. & Geider, K. Characterization of a Viral EPS-Depolymerase, a Potential Tool for Control of Fire Blight. Phytopathology 90, 1263–1268 (2000).</ref> that can hydrolyze the Exopolysaccharide (EPS) forming the biofilm. |
| | | | |
| − | ''Erwinia amylovora'' is gram-negative bacterium in the family Enterobacteria. This bacterium have a similar path of infection as Xylella Fastidiosa, Erwinia amylovora makes its entry into its host xylem synthesis the extracellular polysaccharides (EPS) occluding the xylem vessels of the plant.
| + | As the symptoms observed in plants are the result occlusion of xylem vessels by bacterial biofilm and the accumulation of EPS, |
| | + | we think this may be a way to unblock the vessels. |
| | + | The goal of part of the design isn’t to treat [[Team:Aix-Marseille/Xylella_fastidiosa|''X. fastidiosa'']] or to prevent the infection, but to reduce symptoms. |
| | | | |
| − | This information narrowed my research in the direction of determining enzymes capable of degrading the biofilm of ''Xylella fastidiosa'' rendering it more susceptible to antibiotic, naturally present phages or the natural defence of the plant.
| + | ==EPS Depolymerase design== |
| | | | |
| − | First step was to run a blast using the EPS-Depolymerase from ''Erwinia amylovora'' (Q9G072_9CAUD) to see if an EPS-Depolymerase was present in phages against ''Xylella fastidiosa'', but alas the results where negative but a potential candidate arose.
| + | [[File:T--Aix-Marseille--EPSplasmid-2.png|450px|right|thumb]] |
| | | | |
| − | Putative tail fiber protein/putative EPS Depolymerase (DIBBI_029) is an enzyme of 852 amino acids, from the organism ''Xanthomonas'' phage vB_XveM_DIBBI which contain two function one structural and one pectin lyase. Multiple articles showed that some phages that work on Xanthomonas can work on ''Xylella fastidiosa'' and Vis versa. However, we can't be sure if the gene chosen can work because of the lack of documentation.
| + | As we wanted to limit the number of GMOs in our product decided to purify the enzyme after production in ''E. coli'' using a quick and efficient purification system. |
| | | | |
| − | [[File:T--Aix-Marseille--DEPS-explication-1.png|800px]] | + | The original enzyme used by the phage is very large and the gene is not optimal for a production in ''E. coli''. |
| | + | Thus, to produce large quantities of the enzyme, we decided to optimize the enzyme by taking only the catalytic domain of the original protein and optimizing the gene sequence for production in ''E. coli''. |
| | + | This lead to the creation of the EPS-Depolymerase part : [http://parts.igem.org/Part:BBa_K2255006 BBa_K2255006]. |
| | | | |
| − | The protein is 852 amino acid which translate to 2556 nucleobases, quite big making it difficult to work with so a process of trimming was done. Seeing as the domain we are interested in pectin lyase start from the 139th to 426th amino acid it is possible to remove the excess amino acids without harming the functionality and specificity of the enzyme.
| + | As we wanted to purify the EPS Depolymerase to use it in our treatment, we also engineered a multi-tag. |
| − | A Blast against NR was done to see if this domain is highly conserved and if the excess were not aligned with the the query sequence DIBBI_029. We found a majority of the alignment started with a gap of 78 amino acids in N-terminus and 266 amino acids in C-terminus so we obted to reduce the size of the protein to 510 amino acids a 342 amino acid reduction.
| + | This tag is composed of an oligo-histidine-tag, that binds to Nickel or Cobalt ions, and a Strep-tag that binds to streptavidin separated by a TEV cleavage site. |
| | + | Thus we created the biobrick [http://parts.igem.org/Part:BBa_K2255003 BBa_K2255003]. |
| | | | |
| − | [[File:T--Aix-Marseille--DEPS-explication-2.png|500px]] | + | The two biobricks will be fused using the [http://parts.igem.org/Assembly_standard_25 Rfc25] standard to produce an easilly purifiable enzyme. |
| | + | To get large amounts, we decided to add a strong and constitutive promoter and RBS in ''E.coli'' ([http://parts.igem.org/Part:BBa_K608002 BBa_K608002]). |
| | | | |
| | + | ==References== |
| | | | |
| − | Giving us the sequence present:
| + | <references/> |
| − | | + | |
| − | <code>
| + | |
| − | MQIQEPDGFKYIGRVPSFAALVSVVPEKAGERVIVSGHVAGNDYGGGVFVARAGSVAINDGGTIMPVNNNFYWQRLVEDPGTLDVTHFGAKRDGVTDCATAC
| + | |
| − | LAMWNYTQSLGAGGSMIGIQFPAGEFAVSNIDISANYVGNFRLVGKGVVTTFGYFPATRIKLIGADNQAAFKVQARRSEIANLQIYGQYEVKANTRGFFKNT
| + | |
| − | CVSGQYVHGVNWRSTYTGGPIFDLMDTLDTKFSEFYASYVYGGVIYGVPSGSESGSWDHLTAIELSNFNVQRCYGKQAFDLQKSGQSFIYNGWIEKTDFPGD
| + | |
| − | LSNGQWIIQGLSMEDCVNPLDLTFTRAQLSQINLQGTSALRYDNPDKSRLLSTYEMGRNRVEAYGAQFFGSLSYDYLSSHYRLSNATDKAAWFNLGKMIVTN
| + | |
| − | QNDASRIRFFGANGQASVPSDQGAFDSNNFGGGECLLTLRRVPGTGTRQDCAIEVHGNSPIADIRISRPYENDVEIYVQLKPQCGFVNVSLETSTNSRFDSG
| + | |
| − | </code>
| + | |
| − | | + | |
| − | Our primary cloning medium is the plasmid pSB1C3, we chose RFC 25 as the prefix and suffix for the sequence and the E.coli promoter
| + | |
| − | | + | |
| − | To purify the protein we added a histidine tag at 3’, which enable a chromatography on a nickel column.
| + | |
| − | The prefix and suffix are special sequence designed with restriction sites to flank both ends of the sequence we are trying to modify. the prefix hold two restriction sites for EcoRI and xbaI, the suffix holds SpeI and PstI.
| + | |
| − | | + | |
| − | The sequence was retro translated to obtain the genomic sequence with the E.coli bias.
| + | |
| − | <code>
| + | |
| − | ATGCAGATTCAGGAACCGGATGGCTTTAAATATATTGGCCGCGTGCCGAGCTTTGCGGCGCTGGTGAGCGTGGTGCCGGAAAAAGCGGGCGAACGCGTGATT
| + | |
| − | GTGAGCGGCCATGTGGCGGGCAACGATTATGGCGGCGGCGTGTTTGTGGCGCGCGCGGGCAGCGTGGCGATTAACGATGGCGGCACCATTATGCCGGTGAAC
| + | |
| − | AACAACTTTTATTGGCAGCGCCTGGTGGAAGATCCGGGCACCCTGGATGTGACCCATTTTGGCGCGAAACGCGATGGCGTGACCGATTGCGCGACCGCGTGC
| + | |
| − | CTGGCGATGTGGAACTATACCCAGAGCCTGGGCGCGGGCGGCAGCATGATTGGCATTCAGTTTCCGGCGGGCGAATTTGCGGTGAGCAACATTGATATTAGC
| + | |
| − | GCGAACTATGTGGGCAACTTTCGCCTGGTGGGCAAAGGCGTGGTGACCACCTTTGGCTATTTTCCGGCGACCCGCATTAAACTGATTGGCGCGGATAACCAG
| + | |
| − | GCGGCGTTTAAAGTGCAGGCGCGCCGCAGCGAAATTGCGAAC<span style="background-color:red">CTGCAG</span>ATTTATGGCCAGTATGAAGTGAAAGCGAACACCCGCGGCTTTTTTAAAAACACC
| + | |
| − | TGCGTGAGCGGCCAGTATGTGCATGGCGTGAACTGGCGCAGCACCTATACCGGCGGCCCGATTTTTGATCTGATGGATACCCTGGATACCAAATTTAGCGAA
| + | |
| − | TTTTATGCGAGCTATGTGTATGGCGGCGTGATTTATGGCGTGCCGAGCGGCAGCGAAAGCGGCAGCTGGGATCATCTGACCGCGATTGAACTGAGCAACTTT
| + | |
| − | AACGTGCAGCGCTGCTATGGCAAACAGGCGTTTGAT<span style="background-color:red">CTGCAG</span>AAAAGCGGCCAGAGCTTTATTTATAACGGCTGGATTGAAAAAACCGATTTTCCGGGCGAT
| + | |
| − | CTGAGCAACGGCCAGTGGATTATTCAGGGCCTGAGCATGGAAGATTGCGTGAACCCGCTGGATCTGACCTTTACCCGCGCGCAGCTGAGCCAGATTAAC<span style="background-color:red">CTG
| + | |
| − | CAG</span>GGCACCAGCGCGCTGCGCTATGATAACCCGGATAAAAGCCGCCTGCTGAGCACCTATGAAATGGGCCGCAACCGCGTGGAAGCGTATGGCGCGCAGTTT
| + | |
| − | TTTGGCAGCCTGAGCTATGATTATCTGAGCAGCCATTATCGCCTGAGCAACGCGACCGATAAAGCGGCGTGGTTTAACCTGGGCAAAATGATTGTGACCAAC
| + | |
| − | CAGAACGATGCGAGCCGCATTCGCTTTTTTGGCGCGAACGGCCAGGCGAGCGTGCCGAGCGATCAGGGCGCGTTTGATAGCAACAACTTTGGCGGCGGCGAA
| + | |
| − | TGCCTGCTGACCCTGCGCCGCGTGCCGGGCACCGGCACCCGCCAGGATTGCGCGATTGAAGTGCATGGCAACAGCCCGATTGCGGATATTCGCATTAGCCGC
| + | |
| − | CCGTATGAAAACGATGTGGAAATTTATGTGCAGCTGAAACCGCAGTGCGGCTTTGTGAACGTGAGCCTGGAAACCAGCACCAACAGCCGCTTTGATAGCGGC
| + | |
| − | </code>
| + | |
| − | | + | |
| − | However, this sequence had three restriction sites for the endonuclease PSTI which is in conflict with the endonuclease we will be using to modify and insert the gene in the plasmid. Keeping these sites will cleave the gene in three separate places losing all functionality.
| + | |
| − | The three sites found on the 555th, 855th and 1020th position with the conserved sequence of CTGCAG, CTG code for a leucine and CAG code for a glutamine. Thankfully, the genomic code is redundant so we can modify nucleobases without changing the protein translated. We chose to change the G of CTG to an A giving us CTA this modification did not produce any restriction sites critical to our work eliminating the restriction site for PSTI.
| + | |
| − |
| + | |
| − | Resulting in the sequence as present
| + | |
| − | | + | |
| − | <code>
| + | |
| − | ATGCAGATTCAGGAACCGGATGGCTTTAAATATATTGGCCGCGTGCCGAGCTTTGCGGCGCTGGTGAGCGTGGTGCCGGAAAAA
| + | |
| − | GCGGGCGAACGCGTGATTGTGAGCGGCCATGTGGCGGGCAACGATTATGGCGGCGGCGTGTTTGTGGCGCGCGCGGGCAGCGTG
| + | |
| − | GCGATTAACGATGGCGGCACCATTATGCCGGTGAACAACAACTTTTATTGGCAGCGCCTGGTGGAAGATCCGGGCACCCTGGAT
| + | |
| − | GTGACCCATTTTGGCGCGAAACGCGATGGCGTGACCGATTGCGCGACCGCGTGCCTGGCGATGTGGAACTATACCCAGAGCCTG
| + | |
| − | GGCGCGGGCGGCAGCATGATTGGCATTCAGTTTCCGGCGGGCGAATTTGCGGTGAGCAACATTGATATTAGCGCGAACTATGTG
| + | |
| − | GGCAACTTTCGCCTGGTGGGCAAAGGCGTGGTGACCACCTTTGGCTATTTTCCGGCGACCCGCATTAAACTGATTGGCGCGGAT
| + | |
| − | AACCAGGCGGCGTTTAAAGTGCAGGCGCGCCGCAGCGAAATTGCGAAC<span style="background-color:red">CTACAG</span>ATTTATGGCCAGTATGAAGTGAAAGCGAAC
| + | |
| − | ACCCGCGGCTTTTTTAAAAACACCTGCGTGAGCGGCCAGTATGTGCATGGCGTGAACTGGCGCAGCACCTATACCGGCGGCCCG
| + | |
| − | ATTTTTGATCTGATGGATACCCTGGATACCAAATTTAGCGAATTTTATGCGAGCTATGTGTATGGCGGCGTGATTTATGGCGTG
| + | |
| − | CCGAGCGGCAGCGAAAGCGGCAGCTGGGATCATCTGACCGCGATTGAACTGAGCAACTTTAACGTGCAGCGCTGCTATGGCAAA
| + | |
| − | CAGGCGTTTGAT<span style="background-color:red">CTACAG</span>AAAAGCGGCCAGAGCTTTATTTATAACGGCTGGATTGAAAAAACCGATTTTCCGGGCGATCTGAGC
| + | |
| − | AACGGCCAGTGGATTATTCAGGGCCTGAGCATGGAAGATTGCGTGAACCCGCTGGATCTGACCTTTACCCGCGCGCAGCTGAGC
| + | |
| − | CAGATTAAC<span style="background-color:red">CTACAG</span>GGCACCAGCGCGCTGCGCTATGATAACCCGGATAAAAGCCGCCTGCTGAGCACCTATGAAATGGGCCGC
| + | |
| − | AACCGCGTGGAAGCGTATGGCGCGCAGTTTTTTGGCAGCCTGAGCTATGATTATCTGAGCAGCCATTATCGCCTGAGCAACGCG
| + | |
| − | ACCGATAAAGCGGCGTGGTTTAACCTGGGCAAAATGATTGTGACCAACCAGAACGATGCGAGCCGCATTCGCTTTTTTGGCGCG
| + | |
| − | AACGGCCAGGCGAGCGTGCCGAGCGATCAGGGCGCGTTTGATAGCAACAACTTTGGCGGCGGCGAATGCCTGCTGACCCTGCGC
| + | |
| − | CGCGTGCCGGGCACCGGCACCCGCCAGGATTGCGCGATTGAAGTGCATGGCAACAGCCCGATTGCGGATATTCGCATTAGCCGC
| + | |
| − | CCGTATGAAAACGATGTGGAAATTTATGTGCAGCTGAAACCGCAGTGCGGCTTTGTGAACGTGAGCCTGGAAACCAGCACCAAC
| + | |
| − | AGCCGCTTTGATAGCGGC
| + | |
| − | </code> | + | |
| − |
| + | |
| − | If all well and done, we should be able to clone the gene and recuperate the protein with a Molecular weight of 55.81 kD that is 510 amino acids without taking into account the histidine tag.
| + | |
EPS Depolymerase
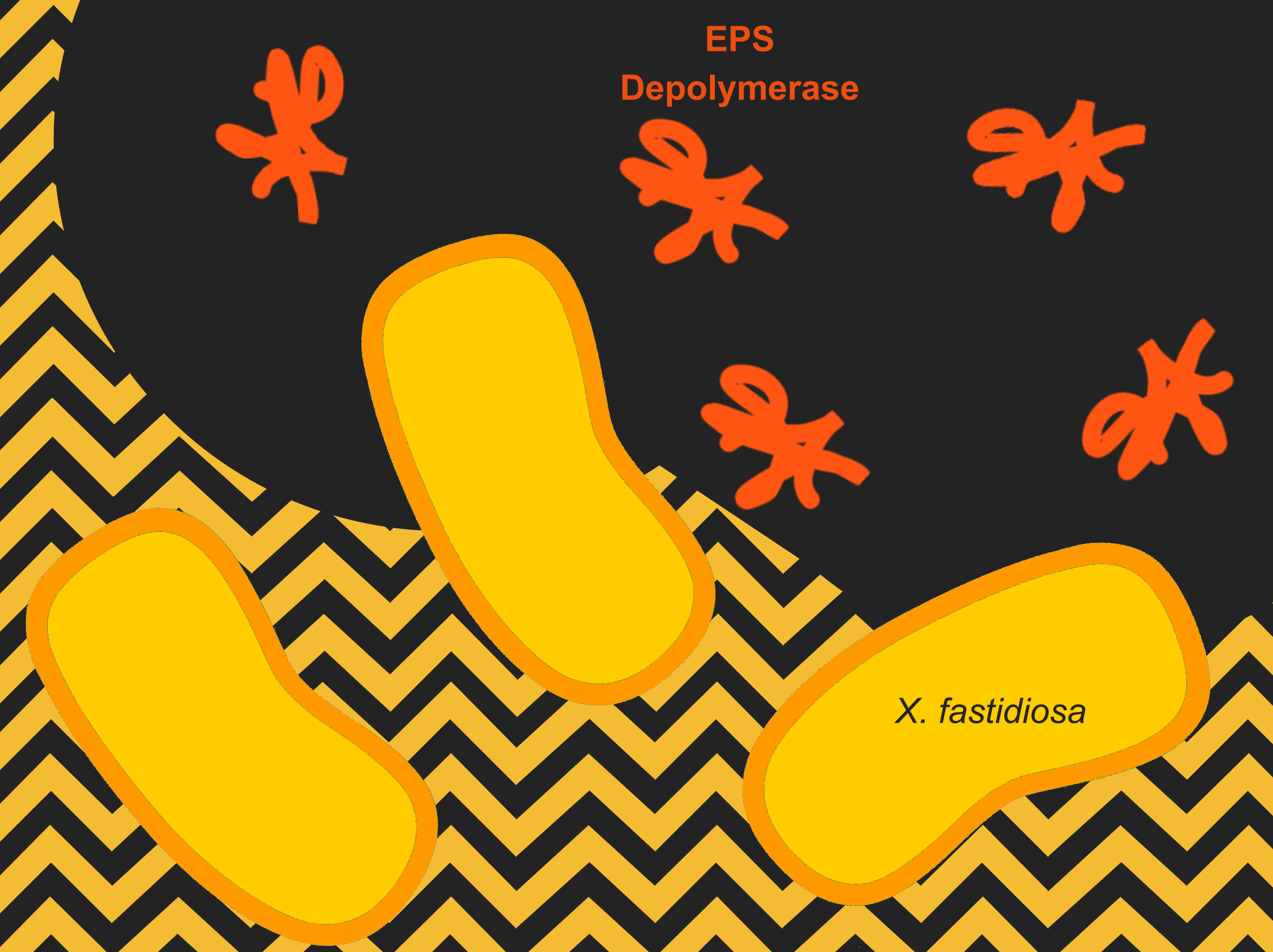
EPS Depolymerase process to clean the xylem vessel from EPS.
To fight Xylella fastidiosa we searched for natural solutions.
Some phages have a devious way to attack bacteria, digesting the biofilm so that they can their target more easily [1].
We decided to try to use this approach to attack Xylella fastidiosa.
We found an enzyme called EPS-depolymerase [2] that can hydrolyze the Exopolysaccharide (EPS) forming the biofilm.
As the symptoms observed in plants are the result occlusion of xylem vessels by bacterial biofilm and the accumulation of EPS,
we think this may be a way to unblock the vessels.
The goal of part of the design isn’t to treat X. fastidiosa or to prevent the infection, but to reduce symptoms.
EPS Depolymerase design
As we wanted to limit the number of GMOs in our product decided to purify the enzyme after production in E. coli using a quick and efficient purification system.
The original enzyme used by the phage is very large and the gene is not optimal for a production in E. coli.
Thus, to produce large quantities of the enzyme, we decided to optimize the enzyme by taking only the catalytic domain of the original protein and optimizing the gene sequence for production in E. coli.
This lead to the creation of the EPS-Depolymerase part : [http://parts.igem.org/Part:BBa_K2255006 BBa_K2255006].
As we wanted to purify the EPS Depolymerase to use it in our treatment, we also engineered a multi-tag.
This tag is composed of an oligo-histidine-tag, that binds to Nickel or Cobalt ions, and a Strep-tag that binds to streptavidin separated by a TEV cleavage site.
Thus we created the biobrick [http://parts.igem.org/Part:BBa_K2255003 BBa_K2255003].
The two biobricks will be fused using the [http://parts.igem.org/Assembly_standard_25 Rfc25] standard to produce an easilly purifiable enzyme.
To get large amounts, we decided to add a strong and constitutive promoter and RBS in E.coli ([http://parts.igem.org/Part:BBa_K608002 BBa_K608002]).
References
- ↑ Vandenbergh, P. A., Wright, A. M. & Vidaver, A. K. Partial Purification and Characterization of a Polysaccharide Depolymerase Associated with Phage-Infected Erwinia amylovora. Appl. Environ. Microbiol. 49, 994–996 (1985)
- ↑ Kim, W. S. & Geider, K. Characterization of a Viral EPS-Depolymerase, a Potential Tool for Control of Fire Blight. Phytopathology 90, 1263–1268 (2000).

