Difference between revisions of "Team:UNOTT/Experiments"
MattAFrench (Talk | contribs) |
|||
| (15 intermediate revisions by 3 users not shown) | |||
| Line 251: | Line 251: | ||
<div class="whiteboxmargin"> | <div class="whiteboxmargin"> | ||
<h1>Why</h1> | <h1>Why</h1> | ||
| − | <p> | + | <p>In our experiment, the production of differentially identifiable expression patterns is paramount. A library of promoters with robust expression patterns was required to achieve this. To further modulate the expression patterns, a promoter targeted short guide RNA (sgRNA)-Cas9 repression system was devised. These sgRNAs have a 20bp SEED region homologous to the promoter between the -10 and -35 box, a deactivated Cas9 associates with the sgRNA and is directed to the promoter through it’s homology, this will inhibit the RNA polymerase as it’s very large and bind with a high affinity. The strength of inhibition is proportional to the binding afinity of the SEED region. <br> |
| + | A two vector based system was decided upon to express this system. One low copy vector will express the catalytically inactive Cas9 protein, promoters and a downstream reporter; the other will contain the sgRNAs and a high copy origin of replication (ori). Confining the sgRNAs to a separate vector is advantageous in that it can be easily substituted out for another vector, potentially detailing sgRNAs with a differing affinity to the promoter. This interchangeability can facilitate a ‘plug and play’ system to produce a multitude of expression levels with ease. | ||
| + | </p> | ||
<h1>How</h1> | <h1>How</h1> | ||
| − | <p> Refer to step 2 for plasmid construction. | + | <p> Refer to step 2 for plasmid construction.To select for a dual transformation, each vector backbone requires separate antibiotic resistances. Consequently, the high copy vector (containing sgRNAs) was given an erythromycin resistant cassette, the low copy backbone was given a Chloramphenicol resistance cassette. Each vector also required a gram-negative compatible ori. High copy ColE1 was present on the sgRNA vector to drive high expression and promoter inhibition. A low copy pSC101 ori was initially given to the other, as dCas9 is toxic in high concentrations. |
| + | Another set of sgRNAs were designed to have no inhibitory activity, termed sgRNA0s. The ‘seeds’ were not homologous to any of the promoter, whereas the other set were 100% homologous. This will act as a control, to signify how the expression will differ when inhibition is occurring. It will also serve later to increase the pool of possible expression patterns when we come to the random ligation experiments. These were cloned using traditional type II restriction cloning with BsaI sites.<br><br> | ||
| + | To construct the sgRNAs, primer dimers were PCR amplified to generate the initial fragments of the sgRNA, it’s promoter, and terminator; also with their complimentary BsaI sites. After digestion with BsaI, the fragments were linearly ligated to produce promoter-sgRNA-terminator ‘bricks’. These were then amplified using a set of ‘universal’ primers designed to be capable of amplifying all of our ‘bricks’, these primers were complementary to a multiple cloning site (MCS) flanking each brick we produced. These sgRNA bricks were then digested with this MCS and ligated into their associated high copy backbone. | ||
| + | </p> | ||
<h1>Results</h1> | <h1>Results</h1> | ||
| − | <p> | + | <p>For the CRISPRi results check step 6 and the lab-book for more in detail descriptions of the prodution of the gRNA and gel images.</p> |
| Line 291: | Line 296: | ||
<p>After this, the same method as above was used for the final dCas9 including vector. However with an addition of dCas9 in the final ligation phase. In this way two promoter libraries were produced.</p> | <p>After this, the same method as above was used for the final dCas9 including vector. However with an addition of dCas9 in the final ligation phase. In this way two promoter libraries were produced.</p> | ||
<h1>Results</h1> | <h1>Results</h1> | ||
| − | <p> | + | <p>For results check steps 3 and 4.</p> |
| Line 312: | Line 317: | ||
<div class="whiteboxmargin"> | <div class="whiteboxmargin"> | ||
<h1>Why</h1> | <h1>Why</h1> | ||
| − | <p> | + | <p> |
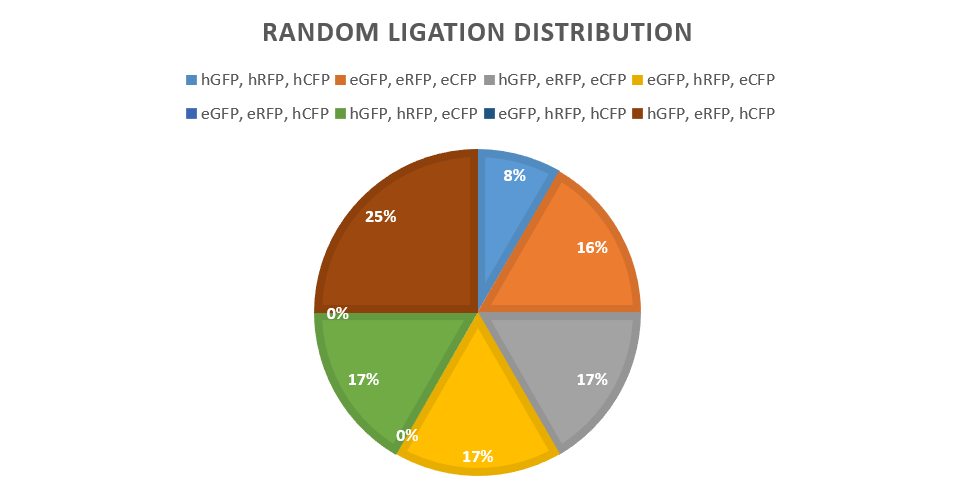
To establish the relative strength of every single promoter, each of them must be tested and their expression level measured throughout the bacterial growth. This will provide data to demonstrate it is possible to differentiate between them, proving that our key will work effectively by having identifiable spectral states. Getting unique spectral profiles is a cause of concern as the excitation and emission spectra of RFP, CFP and GFP overlap somewhat and cross excitation will occur, perhaps masking CFP’s emissions and reducing the potential for separate keys to be identifiable. To fully address this, a promoter library was constructed to assess the promoter’s strength. | To establish the relative strength of every single promoter, each of them must be tested and their expression level measured throughout the bacterial growth. This will provide data to demonstrate it is possible to differentiate between them, proving that our key will work effectively by having identifiable spectral states. Getting unique spectral profiles is a cause of concern as the excitation and emission spectra of RFP, CFP and GFP overlap somewhat and cross excitation will occur, perhaps masking CFP’s emissions and reducing the potential for separate keys to be identifiable. To fully address this, a promoter library was constructed to assess the promoter’s strength. | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
</p> | </p> | ||
<h1>How</h1> | <h1>How</h1> | ||
| − | <p> | + | <p>This growth curve was carried as described in week 15 of the lab book, please refer for full details. To summarise, overnight cultures of each specimen were inoculated into a new batch of media. These were then grown at 37°C for 24 hours, with measurements of OD600nm absorbance and fluorescence carried at 0, 2, 4, 6, and 24 hours. The promoters were paired with a downstream RFP reporter gene.</p> |
<h1>Results</h1> | <h1>Results</h1> | ||
| − | <p><img src="https://static.igem.org/mediawiki/2017/3/30/T--UNOTT--Promoterpoolstrength.png"> | + | <p><table> |
| − | <img src="https://static.igem.org/mediawiki/2017/3/30/T--UNOTT--Promoterpoolstrength.png"><br>Figure 1 illustrates the strengths of each set of strong (SP) and weak (WP) RBS promoters, the first graph being the SPs 1, 2, 3, 4, 5, and E. E is a null promoter which will produce no expression, providing an additional control like sgRNA0 is for the gRNA parts design. It will help making sure no unspecific interference happens when no promoter is present. The second graph shows W1-4. It is clear there are some extremely different expression levels. SP3 and SP4 are quite similar with peaks of 5500-5800 a.u. at 24 hours; it would be hard to discern between them, but these are significantly the strongest promoters and would be useful as a ‘maximum’ expression promoter. SP1 and W4 are also quite comparable with peak around 3000 a.u., these would be considered intermediate/strong promoters. W3, W1, SP2 and perhaps SP5 are all relatively similar with peaks from 900-1700 a.u. at 24 hours. These would be considered weak promoters, they would be useful in a triple reporter experiment to act as the separate targets for the sgRNAs and see if they have distinct inhibited states. W2 is by far the weakest active promoter, with a peak at >500 it would be classed as the weakest promoter, perhaps serving as a minimum activation promoter. SE as expected produced no expression, and can be concluded it will work as a negative control or null promoter. | + | <tr><img src="https://static.igem.org/mediawiki/2017/3/30/T--UNOTT--Promoterpoolstrength.png"></tr> |
| + | <tr><img src="https://static.igem.org/mediawiki/2017/3/30/T--UNOTT--Promoterpoolstrength.png"></tr> | ||
| + | <tr><th>Figure 1. This graph shows the relative fluorescent intesnity of strong and weak RBS promoters SP1-E and WP1-4. These were grown for a 24 hour period and measurements were taken at 0, 2, 4, 6, 9, and 24 hours.</th></tr></table> | ||
| + | <br>Figure 1 illustrates the strengths of each set of strong (SP) and weak (WP) RBS promoters, the first graph being the SPs 1, 2, 3, 4, 5, and E. E is a null promoter which will produce no expression, providing an additional control like sgRNA0 is for the gRNA parts design. It will help making sure no unspecific interference happens when no promoter is present. The second graph shows W1-4. It is clear there are some extremely different expression levels. SP3 and SP4 are quite similar with peaks of 5500-5800 a.u. at 24 hours; it would be hard to discern between them, but these are significantly the strongest promoters and would be useful as a ‘maximum’ expression promoter. SP1 and W4 are also quite comparable with peak around 3000 a.u., these would be considered intermediate/strong promoters. W3, W1, SP2 and perhaps SP5 are all relatively similar with peaks from 900-1700 a.u. at 24 hours. These would be considered weak promoters, they would be useful in a triple reporter experiment to act as the separate targets for the sgRNAs and see if they have distinct inhibited states. W2 is by far the weakest active promoter, with a peak at >500 it would be classed as the weakest promoter, perhaps serving as a minimum activation promoter. SE as expected produced no expression, and can be concluded it will work as a negative control or null promoter. | ||
In conclusion, it is promising that we have 5 distinct states of expression with maximal, strong, weak, minimal, and off. There is also the opportunity to have SP2, W1, W2 and maybe SP5 to act as a way to discern how effective the sgRNA:dCas9 inhibition, owing to their similar expression patterns. | In conclusion, it is promising that we have 5 distinct states of expression with maximal, strong, weak, minimal, and off. There is also the opportunity to have SP2, W1, W2 and maybe SP5 to act as a way to discern how effective the sgRNA:dCas9 inhibition, owing to their similar expression patterns. | ||
</p> | </p> | ||
| − | + | ||
</div> | </div> | ||
</div> | </div> | ||
| Line 409: | Line 412: | ||
<h1>Results</h1> | <h1>Results</h1> | ||
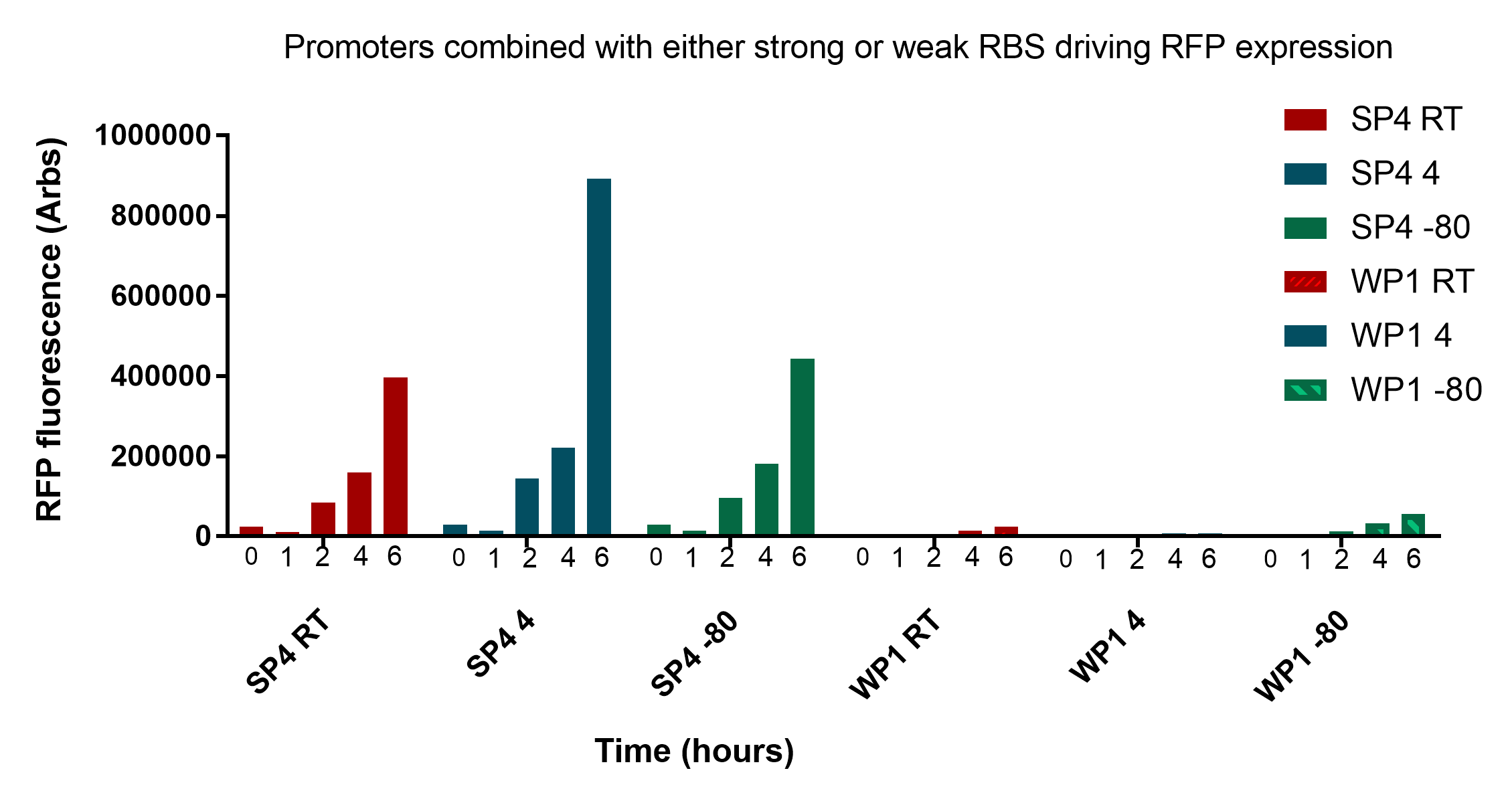
<p>The results presented in figure 1 show that the two promoters, SP4 and WP1, are clearly distinct from each other at every stage of revival and storage time. This information is important as we will need to calibrate the thresholds for identification accordingly. The data also shows that storing the cells for two weeks at different temperatures has no to little effect on the reporter expression.</p> | <p>The results presented in figure 1 show that the two promoters, SP4 and WP1, are clearly distinct from each other at every stage of revival and storage time. This information is important as we will need to calibrate the thresholds for identification accordingly. The data also shows that storing the cells for two weeks at different temperatures has no to little effect on the reporter expression.</p> | ||
| − | <img src="https://static.igem.org/mediawiki/2017/4/40/T--UNOTT--SP4WP1RFP.png"> | + | <table><tr><img src="https://static.igem.org/mediawiki/2017/4/40/T--UNOTT--SP4WP1RFP.png"></tr> |
| − | <p class="imgcaption"><b>Figure 1:</b> Graph of strong promoter 4 and weak promoter 1 transformed cells RFP fluorescence assay after freeze-drying revival two weeks and three weeks after samples were freeze dried. </p> | + | <p class="imgcaption"><tr><th><b>Figure 1:</b> Graph of strong promoter 4 and weak promoter 1 transformed cells RFP fluorescence assay after freeze-drying revival two weeks and three weeks after samples were freeze dried.</th></tr></table> </p> |
<br> | <br> | ||
| − | <p>However, there is a notable decease in fluorescence from 2 to 3 weeks at room temperature, especially pronounced at 4 and 6 hour after revival.. This problem may be mitigated in that the drop offs seem proportional to both WP1 and SP4, so the ratios of expression may remain the same. Having said this, the differences in expression would be within the bounds of the thresholds set to compare the two, as even with the changes they would still be identifiable. One might argue that the need to set so many time and storage dependent thresholds makes the system unnecessary complicated. To minimise the described variables that need to be taken into consideration keys stored at room temperature should be replaced after two weeks. Keys which require long term storage should be stored at - | + | <p>However, there is a notable decease in fluorescence from 2 to 3 weeks at room temperature, especially pronounced at 4 and 6 hour after revival.. This problem may be mitigated in that the drop offs seem proportional to both WP1 and SP4, so the ratios of expression may remain the same. Having said this, the differences in expression would be within the bounds of the thresholds set to compare the two, as even with the changes they would still be identifiable. One might argue that the need to set so many time and storage dependent thresholds makes the system unnecessary complicated. To minimise the described variables that need to be taken into consideration keys stored at room temperature should be replaced after two weeks. Keys which require long term storage should be stored at -80°C.</p> |
<br> | <br> | ||
<p>Interestingly, as predicted there is a substantial amount of fluorescence detectable 2 hours after revival, suggesting that a bacterial key could possibly be identifiable with little waiting period. However, they are undoubtedly more pronounced at 4 and 6 hours which would likely to be a more accurate time to measure.</p> | <p>Interestingly, as predicted there is a substantial amount of fluorescence detectable 2 hours after revival, suggesting that a bacterial key could possibly be identifiable with little waiting period. However, they are undoubtedly more pronounced at 4 and 6 hours which would likely to be a more accurate time to measure.</p> | ||
| + | <p><table><tr><img src="https://static.igem.org/mediawiki/2017/f/f4/Groningen_data_revival.jpeg";></tr> | ||
| + | <tr><th> Figure 2. This graph represents data kindly given to us by the University of Groningen. It details the revival of the two samples containing strong RBS promoter 4 and weak RBS promoter 1 with a RFP reporter. These were grown for an 6 hour period and fluorescence measurements were taken at 0, 1, 2, 4, and 6 hours. One sample each was stored at either room temperature, 4<sup>o</sup>C, or -80<sup>o</sup>C.</th><tr></table></p><br> | ||
| + | <p> | ||
| + | In figure 2, samples were transported to the University of Groningen through the postal courier service, so the samples at -80<sup>o</sup>C would not have been at this temperature for the entire duration up until measuremment. With this in mind, it still appears what was discussed before still holds true. </p> | ||
<br> | <br> | ||
| − | <p>In conclusion, these results are very promising and show that cells remain viable and reliably express the reporter for up to three weeks at convenient storage conditions necessary for our key to work in a practical sense. </p> | + | <p>In conclusion, these results are very promising and show that cells remain viable and reliably express the reporter for up to three weeks at convenient storage conditions necessary for our key to work in a practical sense. The two promoters SP4 and WP1 are very distinct and easily discernible. There still remains a significant amount of viable bacteria regardless of storage conditions and their fluorescence intensity is very comparable at all stages of growth. It is worth noting that measurements were taken at 0 and 1 hours in figure 2, and these showed minimal fluorescent intensity, suggesting that further optimisation needs to occur to achieve 0-1 hour readings. For details on the revival protocol, download <a style="font-weight:bold;" href="https://static.igem.org/mediawiki/2017/9/9d/Revival_protocol.docx">here.</a></p> |
<br> | <br> | ||
<p>For future research, this process could be optimised to produce even more reproducible results and yield a more robust identification process. Distinct 0 hour expression levels might also be possible, greatly increasing the potential of <i>Key. coli</i> becoming a common security system. Multiple reporters should be the next focus to strengthen the security of the key. All reporters will need to be tested for their stability and reproducibility. </p> | <p>For future research, this process could be optimised to produce even more reproducible results and yield a more robust identification process. Distinct 0 hour expression levels might also be possible, greatly increasing the potential of <i>Key. coli</i> becoming a common security system. Multiple reporters should be the next focus to strengthen the security of the key. All reporters will need to be tested for their stability and reproducibility. </p> | ||
| + | <br> | ||
| + | <img src="https://static.igem.org/mediawiki/2017/9/91/T--UNOTT--GroningenData.png" style="width:100%;height:auto;"> | ||
| + | <p class="imgcaption"><tr><th><b>Figure 3:</b> Testing the affect of storage time at various storage temperatures (Room temperature, 4°C and -80°C) on the fluorescence of revived cells expressing strong or weak RFP expression</th></tr></table></p> | ||
<br> | <br> | ||
<br> | <br> | ||
Latest revision as of 03:59, 2 November 2017

EXPERIMENTS:
STEP 1: Create guideRNA Plasmid

STEP 2: Create Reporter Plasmid



STEP 3: Promoter Library


STEP 4: Random Ligations

STEP 5: Freeze Drying & Revival



STEP 6: CRISPRi & gRNA Efficiency


![]()






