Team:UNOTT/Experiments

EXPERIMENTS:
STEP 1: Create guideRNA Plasmid

Why
We first produced our gRNA plasmids in order to stop expression of targeted fluorescent proteins on our reporter plasmid.
How
Results
STEP 2: Create Reporter Plasmid

Why
To create a stepwise and logical order of production, a reporter plasmid was constructed firstly, out of only various promoter types, mRFP and terminators, to allow for testing. This was then escalated to include dCas9 for the final experiment.
How

Each individual Promoter-Reporter-Terminator brick contains interchangeable parts. The three parts are linked together with Bsa1 sites so that there is no preference for any part when ligating together. This allows randomness to be added later. This method is used also for the construction of Promoter-gRNA-Terminator bricks so that this could be randomised in the future. The bricks are then flanked by a prefix and suffix, and these are flanked by restriction sites ABCD on either end. Digestion of bricks with A+B, B+C, and C+D allows any brick to be placed in any position within the plasmid but it would be pre-determined. This means that the no one promoter-reporter-terminator brick would be limited to one specific place in the plasmid, which allows another level of randomness in assembly as we would not know which reporter was being placed where, which could also affect expression levels.
The arrangement of the DCBA restriction system means that any brick can be placed in any position in the plasmid which allows expansion of possibilities whilst maintaining randomness of insertion later. Bricks can be joined together via amplifying each randomly assembled brick through common amplification sites and then cutting them using a set of restriction enzymes which give each plasmid a specific order of bricks, depending on which are cut and then ligated together.

This was then used for the promoter library.
After this, the same method as above was used for the final dCas9 including vector. However with an addition of dCas9 in the final ligation phase. In this way two promoter libraries were produced.
Results
Found in promoter library section.
STEP 3: Promoter Library

Why
A library of promoters with variant expression capacity was constructed for use with reporter proteins. RFP was used as the reporter for ease of screening.How
Results


STEP 4: Random Ligations

Why
The random ligation of different promoter-protein bricks is an experiment to test the viability of a brownian motion driven random ligation process. This random ligation is the basis for the unpredictability of a large scale key design process. The experiment hoped to produce a random mixture of fluorescent protein expression levels. This random mixture of multiple proteins in a single vector would then show the viability of the Key.coli restriction enzyme-ligation process for achieving unpredictability for keys.
How
The highest and lowest performing promoters were chosen to give the most easily visible result. Promoter E and Promoter 4. Promoter 4 gives a high expression of fluorescent proteins, as shown by our promoter library findings. The promoters were then attached to each reporter protein CFP, RFP and GFP via BSAI digestion and ligation (creating no scar sites) along with a terminator to form six "brick" variants. After amplification of the bricks produced via PCR, seven products combinations were ligated to a low copy backbone in the pattern of red, green and then blue (fluorescent protein) consistently through controlled use of the DCBA digestion site. In addition to a set of "random ligations". Each ligation has only one ligation slot (due to availability of restriction cut sites) per reporter type/colour, leaving random chance to produce a combination of all the possible variants in the random ligations where multiple promoters are available.
e= empty promoter, h= high expression promoter
These bricks were ligated with the backbone as follows:
This produced a control for each reporter fluorescent protein in isolation. It also produced a control for all reporters on either highest expression or lowest expression. This finally also produced a random set of colonies for isolation (after transformation) for comparison and test for true randomness.
Results
Twelve colonies were picked from the pilot study of random ligation, which gave the following distribution: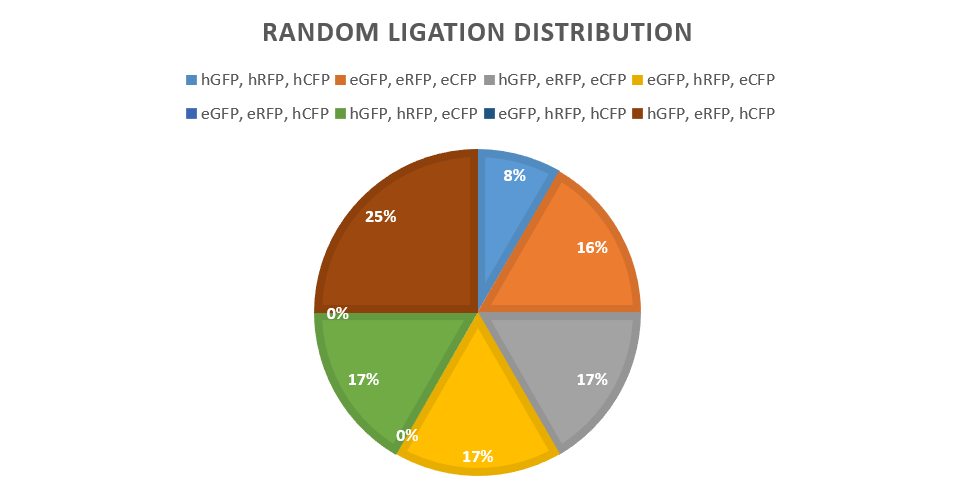
STEP 5: Freeze Drying & Revival

Why
For Key. coli to work as intended and not deteriorate we need need to things to occur:
- The E. coli cells must be kept inactive so that nutrients is not depleted causing the transformed cells to die
- The E. coli cells must be able to be activated after inactivation to allow the fluorescent genes to be expressed to give the key its unique fluorescent code which will allow access to the appliance.
How
To accomplish this, we chose to freeze dry the cells within the key. Click here for out protocol for freeze-drying cells.
Results

Figure 1: Graph of strong promoter 4 and weak promoter 1 transformed cells RFP fluorescence assay after freeze-drying revival two weeks and three weeks after samples were freeze dried.
In our results for freeze-dried cell revival, seen in Figure 1, we can show that storage temperatures do not have an effect on the revival of cells. For strong promoter 4 (SP4), at timepoint of 4 hours and 6 hours the storage temperature does seem to have a negative affect on the relative RFP fluorescence. However our sample size is very small meaning further assays would be needed to confirm this.
STEP 6: CRISPRi & gRNA Efficiency

Why
In our experiment, the production of differentially identifiable expression patterns is paramount. A library of promoters with robust expression patterns was generated to achieve this (step 3 to 5). To further modulate the expression patterns, a promoter targeted short guide RNA (sgRNA)-dcas9 repression system was devised. Using such a system will add a third expression level to the already existing ON and OFF states of our system, expanding the options to xxx?
The sgRNAs have a 20bp ‘seed’ region homologous to the promoters (used in step 3) between the -10 and -35 box, which will form a complex with a deactivated Cas9. The complex will be directed to the promoter through the homology between promoter sequence and the sgRNA. Binding of this very big RNA-protein complex to the promoter sequence will inhibit recognition and binding of RNA polymerase to the promoter region due to allosteric hindrance. The strength of inhibition is related to the binding affinity of the complex to the target DNA. Ideally, each promoter will have its own unique sgRNA which only inhibits the intended promoter to prevent cross interference.
How
To exemplify that the presence of a repression system would allow an increase in possible key outputs so we decided to start with a simple proof of function experiment. Therefore, we decided to go for a two plasmid based system.
One low copy vector was used to express both the catalytically inactive Cas9 protein and the RFP reporter, fused to any of the five promoters previously tested in step 3. Using a low-copy plasmid for the expression of reporter and dCas9 was a precautionary measure, as high levels of dCas9 can be toxic to the cells due to unspecific binding to other regions of the genome. Starting the experiments with only one reporter would allow us to easily establish how well each sgRNA represses the complementary promoter. The second plasmid was comprised of the sgRNAs and a high copy origin of replication (ori). Confining the sgRNAs to a separate vector is advantageous in that it can be easily substituted out for another vector, potentially detailing sgRNAs with a differing affinity to the promoter. This interchangeability can facilitate a ‘plug and play’ system to produce a multitude of expression levels with ease.
To select for a dual transformation, each vector backbone requires separate antibiotic resistances. Consequently, the high copy vector (containing sgRNAs) was given an chloramphenicol resistant cassette, the low copy backbone was given a erythromycin resistance cassette.
As a control, a sgRNA 0 was added to the library. The ‘SEED’ region of this sgRNA is not homologous to any of the promoters. This will act as a control, to signify how the expression will differ when inhibition is occurring. It will also serve later to increase the pool of possible expression patterns when we come to the random ligation experiments . These were cloned using traditional type II restriction cloning with BsaI sites.
To construct the sgRNAs, primer dimers were PCR amplified to generate the initial fragments of the sgRNA, it’s promoter, and terminator; also with their complimentary BsaI sites. After digestion with BsaI, the fragments were linearly ligated to produce promoter-sgRNA-terminator ‘bricks’. These were then amplified using a set of ‘universal’ primers designed to be capable of amplifying all of our ‘bricks’, these primers were complementary to a multiple cloning site(MCS) flanking each brick we produced. These sgRNA bricks were then digested using this MCS and ligated into their associated high copy backbone.
Results

The above graph shows characterization of the sgRNAs. As you can see, gRNA3, 4 and 5 are repressing well. gRNA 1 and 2 have no repression, and PE shows that there is very little expression as a background level.
![]()






