Team:CCA San Diego/Model

protein and chemical modelling
Powered by Pymol and Swiss-Model
Background
There are several enzymes involved in the PAH upper catabolic pathway. The aerobic degradation of aromatic compounds is typically initiated by oxygenases that catalyze the incorporation of two oxygen atoms into the aromatic ring followed by a dehydrogenation reaction catalyzed by dehydrogenases. Theses enzymes are designated RHDO (ring hydroxylating dioxygenases). The dioxygenases responsible for the first step in the aerobic oxidation of lower molecular weight aromatic hydrocarbons, e.g. naphthalene, biphenyl, benzene, and certain other aromatic compounds, share many similarities with genes for which the sequence is known. In contrast, there is little information about bacterial genes encoding proteins for the degradation of higher molecular weight PAH such as phenanthrene and fluorene.
RHDO enzymes are typically soluble, multi-component systems that are usually made up of two or three separate proteins arranged in a short electron transport chain where electrons are moved to the catalytic terminal oxygenase including a ferredoxin, a ferredoxin reductase and an iron sulfur protein. The iron-sulfur protein is made up of an alpha and beta subunit. The alpha subunits contain the catalytic site structured as a pocket to favor the interaction with a hydrophobic PAH.
Phenanthrene PathwayphnAc is a ring hydroxylating dioxygenase alpha subunit that is the catalytic site with a domain exhibiting a deep hydrophobic ligand-binding pocket. phnAd is a ring hydroxylating dioxygenase beta subunit with no known function except for a structural role. Figure 1. Reaction catalyzed by the hydroxylating dioxygenase phnAc/phnAd as the first step of the phenanthrene degradation pathway. |
Fluorene PathwaydbfA1 is an angular dioxygenase large subunit that is the catalytic site . dbfA2 is the angular dioxygenase small subunit (source). Figure 2. Reaction catalyzed by the angular dioxygenase dbfA1/dbfA2 as the first step of the fluorene degradation pathway. |
Modelling project goal
The goal of our modeling project was to further characterize the active sites of the dioxygenase domains of phnAc and dbfA1. To gain insight in this interaction, we have compared their sequences with other dioxygenases for which the crystallization and/or the substrate specificity were determined to assess whether there was a recognizable amino acid pattern specific to each PAH.
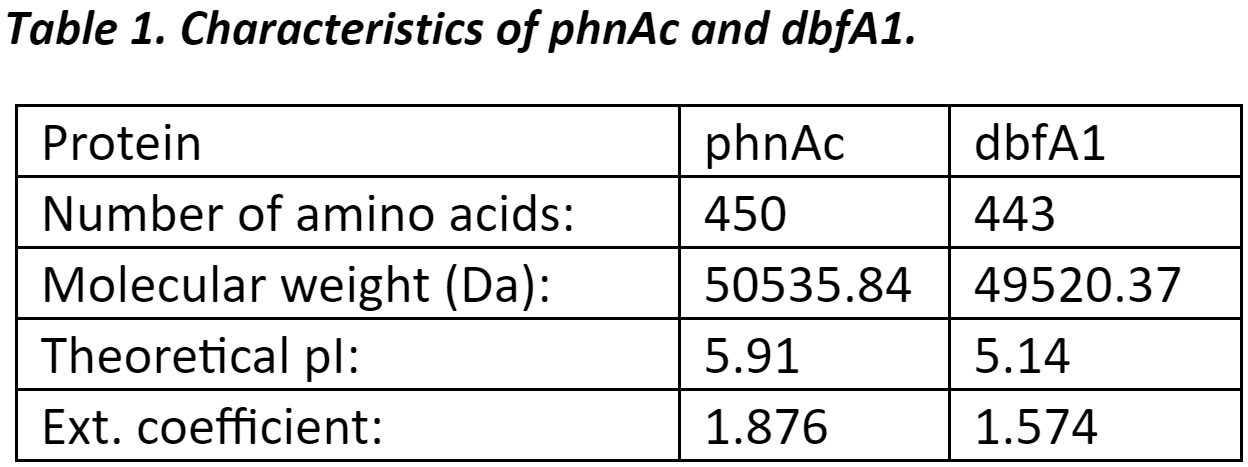
Protein InformationThe protein sequences used in this project were first analyzed to get basic information such as number of amino acids, molecular weight, and theoretical pI (source). |
Protein Modelling
For the modelling, we used SWISS-MODEL, a web-based software that build protein models based on homology by identifying structural template(s), aligning sequences and template structures, building the putative model, and then evaluating the model by statistical analysis. GMQE (Global Model Quality Estimation) is a score between 0 and 1 that estimates the accuracy of a model aligned with a given template. High GMQE reflects higher accuracy. QMEAN is a score that estimates the ‘degree of nativeness’ of the structural features observed in the model. Higher QMEAN indicates agreement between the model structure and experimental structures.
Identification of Amino Acid Residues in the Ligand Binding Pocket Interacting with PAH
To identify the amino acid residues potentially interacting with PAH, we used SWISS-MODEL and PYMOL.
Multiple Sequence Alignment with phnAc (Phenanthrene) and dbfA1 (Fluorene)
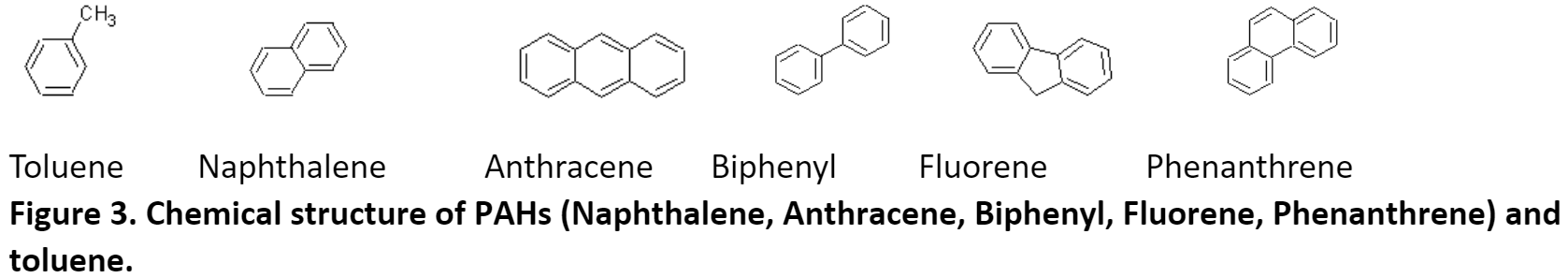
The amino sequence homology between phnAc, dbfA1, and six proteins identified by blast analysis to have similar 3D-domains and using Clustalw. The six proteins listed below have served as the structural model of this enzyme class of dioxygenases which includes naphthalene, toluene, biphenyl, and phenanthrene.

Results for phnAc (Phenanthrene) and dbfA1 (Fluorene)
Amino acid residues of phnAc and dbfA1 were threaded using SWISS-MODEL. The template for the modeling of phnAc was an enzyme for which the co-crystal structure of the protein with its ligand has been experimentally determined. The template for the modeling of dbfA1 was an enzyme with a known crystal. In both cases, the modeling revealed the presence of an active site in a form of a pocket to bind to the polycyclic aromatic hydrocarbon and the iron.
The modeling also revealed that, despite a low (<50%) sequence identity between these dioxygenases, their overall structures was conserved and could superpose. The differences in the active site of the enzymes were more pronounced for toluene and fluorene whereas they were more conserved for naphthalene, phenanthrene, and anthracene. The amino acid residues in the PAH-binding pocket may all be responsible for the catalytic reaction and the orientation/conformation of the PAH inside the pocket. Phenylalanyl (F) and aspartic acid (D) were conserved in this region. The cooperative actions of residues may determine the substrate specificity of the enzyme.
 Figure 4. Modeling of phnAc. This model thread using SWISS_MODEL is based on alignment with naphthalene 1,2-dioxygenase co-crystalized with 1-chloronaphthalene (PDB: 4HJL_A). Structure of the potential active site is shown with naphthalene (grey sticks) docked in the active site and a mononuclear iron (orange sphere). The modeling was considered reasonably accurate with GMQE and QMean scores of 0.78 and -1.41 respectively. |
 Figure 5. Modeling of dbfA1. This model thread using SWISS_MODEL is based on the crystal structure of biphenyl 2,3-dioxygenase from Sphingomonas yanoikuyae B1 (PDB: 2GBW_A). Structure of the potential active site is shown with a mononuclear iron (orange sphere). GMQE and QMean scores were 0.68 and -4.40 respectively. |
 Figure 6.Co-crystal structures (green sticks) of active sites of 2,3-dioxygenase bound to toluene [PDB 3EN1_A, left panel], and naphthalene 1,2-dioxygenase bound to 1-chloronaphthalene [PDB: 4HJL_A, right panel]. The ligands (toluene and 1-chloronaphthalene) are shown in orange and distances between ligands and amino acid residues were measured (yellow lines) to identify amino acid residues inside the ligand-binding pocket in contact with the ligand. These crystal structures were used to model phnAc and dbfA1. |
Conclusion
Further studies involving replacements of specific residues of the substrate-binding pocket by mutagenesis should bring new insights into the role of these residues in the catalytic activity of this class of enzymes. Furthermore, the potential to tailor/engineer the specificity of PAH recognition make the dioxygenases very useful to broaden bioremediation to a larger number of toxic compounds.
 Figure 7. Multiple sequence alignment of catalytic domains of phnAc and dbfA1 with other dioxygenases.The amino acid residues of phnAc, dbfA1, and dioxygenases with known crystal structure were aligned using ClutalWOmega. The amino acid residues determined to be in contact with PAH either by co-crystallization or by modeling are boxed. |
 Figure 8. Alignment of amino acid residues possibly interacting with PAHs in the ligand binding domains of phnAc and dbfA1 and other proteins interacting with PAHs. |




Figure 9. Sequences. Amino acid sequences used in the alignments are listed below. |
Carredano E, Karlsson A, Kauppi B, Choudhury D, Parales RE, Parales JV, Lee K, Gibson DT, Eklund H, Ramaswamy S. Substrate binding site of naphthalene 1,2-dioxygenase: functional implications of indole binding. J Mol Biol. 2000 Feb 18;296(2):701-12. Ferraro DJ, Brown EN, Yu CL, Parales RE, Gibson DT, Ramaswamy S, Structural investigations of the ferredoxin and terminal oxygenase components of the biphenyl 2,3-dioxygenase from Sphingobium yanoikuyae B. BMC Structural Biology20077:10 Friemann, R., Lee, K., Brown, E. N., Gibson, D. T., Eklund, H., & Ramaswamy, S. (2009). Structures of the multicomponent Rieske non-heme iron toluene 2,3-dioxygenase enzyme system. Acta Crystallographica Section D: Biological Crystallography, 65(Pt 1), 24–33. Furusawa Y, Nagarajan V, Tanokura M, Masai E, Fukuda M, Senda T. Crystal structure of the terminal oxygenase component of biphenyl dioxygenase derived from Rhodococcus sp. strain RHA1. J Mol Biol. 2004 Sep 17;342(3):1041-52.
Chemical Modelling
Background
Polycyclic Aromatic Hydrocarbon (high weight molecules) have not been extensively studied and therefore, the pathways and associated degradation genes are not well characterized. In order to understand the nature of the organic reactions taking place during the degradation of PAHs, we had to model the theoretical chain of reactions that would lead to salicylate and phthalate using known chemical catalysts. We predict that the active sites of our biomolecular system will be very similar to the degradation of the chemicals that we have modeled below. The modeling of such pathways allows us to scope out any harmful byproducts (that may kill our cells) or intermediates of our proposed pathways as well as predict any possible deviations for the pathway that may contribute to substantial loss in salicylate or phthalate product, if this were to occur. We also modelled the degradations of the products of our system (salicylate and phthalate) in order to confirm the lack of toxicity this pathway presents to our cells, as well as to confirm its use in carbon-related energy harvesting. We predict that if pyruvate molecules are created along the degradation process, we may be certain that the coded pathway will be used as a way of carbon-energy harvesting by our synthetic E. coli strains and therefore can be used during measurement of cell growth.
 Figure 10. Phenanthrene Pathway. Phenanthrene molecule degrades into salicylate and two pyruvate molecules along a modeled path. |
 Figure 11. Fluorene Pathway. Fluorene molecule degrades into a phthalate, a pyruvate, and an acetaldehyde molecule along a modeled path. |
 Figure 12. Salicylate and Phthalate Pathway. Phthalate molecule degrades into an acetaldehyde and pyruvate molecule in an E. Coli bacterium. Salicylate can be incorporated into the pathway with salicylate hydroxylase. |
 
Figure 13. Substance Reference Sheet. |
Conclusion
The simplest possible pathways were modeled and clearly show the steps to which the organic catabolic degradation can occur along our pathway. Because Salicylate and Phthalate are readily degraded by many cells for energy usage, they act as “intermediates” that allow our synthetic pathways to end at salicylate and phthalate and because of the relatively small numbers of genes possible in a theoretical pathway for fluorene and phenanthrene, biomolecule gene operons associated with these specific organic reactions (some which can combine multiple) were sought after (see pathway data mining). Because there is very small amount of data present on our modified genes, we are not able to isolate the activity of the active sites, but only guess at the possibility of their role in the above mentioned pathways. We also notice that the all PAH degradation pathways used result in simple chemical molecules that may be used by the cell. We predict that the pathways will allow PAHs to be used as metabolic energy for the cell (notice the existence of pyruvate molecules that will eventually lead into the Krebs cycle).

Above is an example of the degradation schematic that helped further our design.
Canyon Crest Academy iGEM 2017 CC; |






