Difference between revisions of "Team:Lethbridge/Design"
| Line 157: | Line 157: | ||
<center> | <center> | ||
<img src="https://static.igem.org/mediawiki/2017/1/17/T--Lethbridge--propur.png" width=750px; height=769px; padding: 30px class="bannerImg" /> | <img src="https://static.igem.org/mediawiki/2017/1/17/T--Lethbridge--propur.png" width=750px; height=769px; padding: 30px class="bannerImg" /> | ||
| − | <br><br>< | + | <br><br> |
| + | <hr class="dividerLine"> | ||
| + | |||
<h2 align= "left"> RNA Purification</h2> | <h2 align= "left"> RNA Purification</h2> | ||
Revision as of 16:28, 31 October 2017

Design Objectives
Our design was guided by the needs of three user groups:
 |
 |
 |
|---|---|---|
| Simple science protocols | Standardized parts | Well characterized and developed system |
| Standard, easy-to-use system | Simplified protocol | Modular tool that can be tailored to the application |
| Modular experiments | Open-source | Open-source |
| Promote good safety practices | Standardized parts | Advancement of cell-free systems |
Resulting in the identification of four core design considerations for our system:

Overview
To ensure the utility of our system, we developed a simplified expression and purification strategy to produce the necessary biomachinery for cell-free synthetic biology.
Our system includes all of the 38 essential TX-TL proteins, the 23S and 16S rRNA, and a tRNA for each amino acid, where tRNAPhe will act as a proof of concept for our novel purification method. In addition, we include the MS2 coat protein which is essential to our tRNA and rRNA purification strategies.

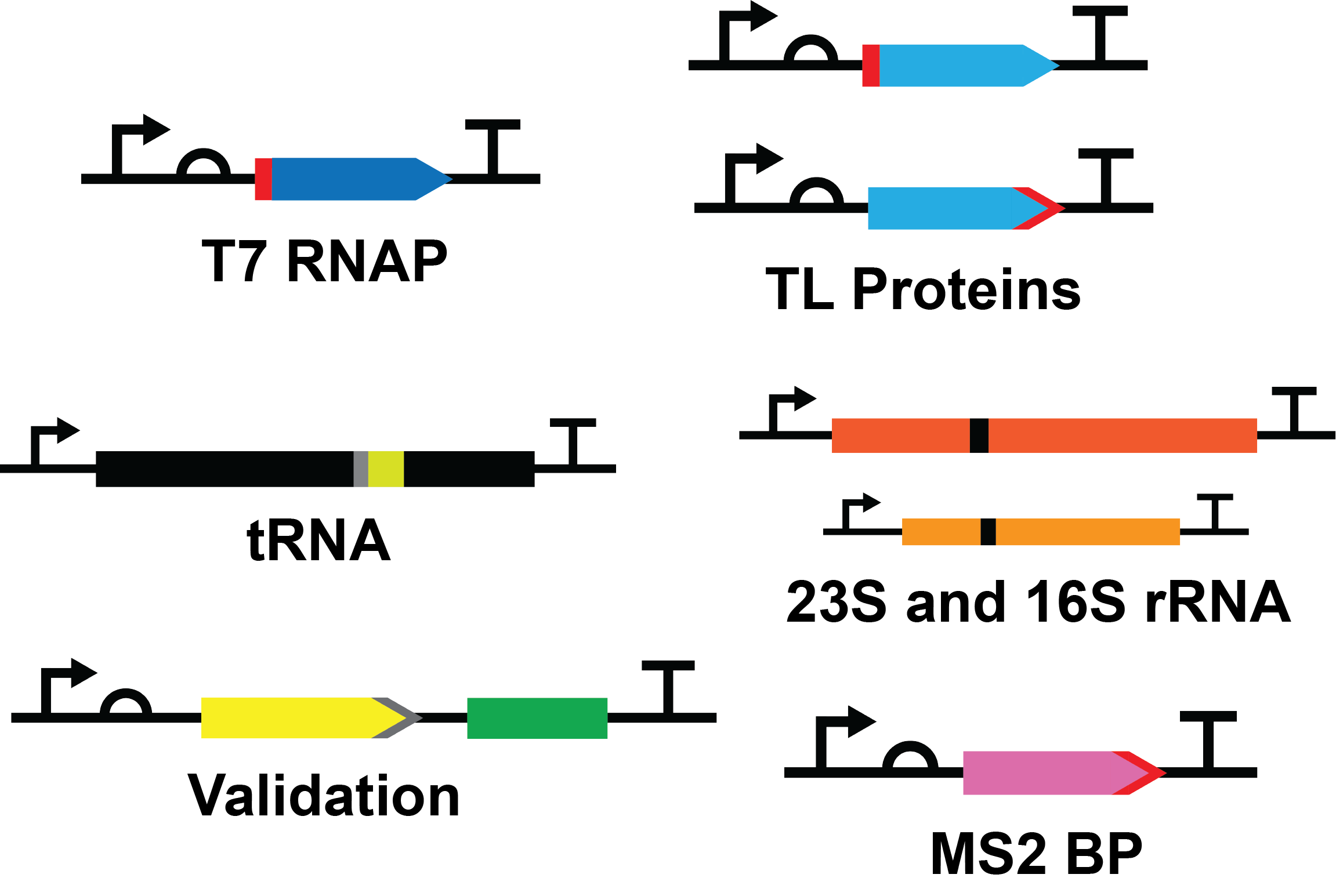
Standard Construct Design
Protein Constructs
Each protein construct consists of a T7 promoter (BBa_I719005), medium ribosome binding site (RBS) (BBa_B0034), protein coding sequence, serine-glycine linker and hexahistidine tag at the N- or C-terminus, and a double terminator (BBa_B0015).
The promoter, RBS and terminator are standard BioBrick parts well-characterized in the registry, providing a good starting point for the initial characterization of our system. Each construct was also codon optimized to allow for efficient expression in E. coli.

RNA Constructs
Each RNA construct consisted of a T7 promoter (BBa_J64997), RNA product, and a double terminator (BBa_B0015).

Purification Strategy
Protein Purification
Transcription and translation factors are expressed with a 6 histidine residue tag to allow for simple purification by nickel sepharose affinity chromatography. The hexahistidine tag is encoded at the N- or C-terminus of each gene based on what was previously described in the literature [1].

RNA Purification
To purify RNA, we take advantage of a previously published purification strategy adapted from the MS2 bacteriophage [2]. It takes advantage of a specific RNA: protein interaction between an RNA hairpin in the MS2 genome and the MS2 coat protein (MS2BP) that forms the viral capsid. The MS2BP has a hexahistidine tag expressed at its C-terminus. This alteration gives the MS2 coat protein dual binding ability. It will have high affinity to nickel as well as to the MS2 hairpin. Thus, tRNA and rRNA expressing an MS2 hairpin can be purified in the same column as the hexahistidine-tagged proteins.

We developed a novel purification system based on a general construct, whereby an RNA of interest will be transcribed with a scar and spacer region, acting as an oligo binding site. A DNA oligo can bind to this site, resulting in a DNA-RNA hybrid, as a target for cleavage by RNase H. This will be followed by two MS2 hairpins, enabling tight binding to the MS2BP for purification.

Our system is designed such that all components can be purified at the same time by nickel sepharose affinity chromatography!

Component Ratios
While the TX-TL system consists of 38 proteins, it requires the function of several proteins more than others. For example, the elongation factors (EF-G, EF-Tu, and EF-Ts) are involved in the addition of each amino acid while the release factors (RF1, RF2, RF3) are involved only when translation has reached the stop codon. As such, the TX-TL system requires a higher concentration of the elongation factors relative to the release factors. Overall the concentration difference between all 38 TX-TL components ranges from 6 ug/mL to 1000 ug/mL. Taking this into consideration we devised several strategies for how we would design our constructs and/or expression strategy to ensure that each protein was purified to the proper final concentration.

The ideal final design of the Next vivo system was to have each TX-TL component expressed from a single plasmid or bacterial artificial chromosome (BAC) with all genes under different promoters and ribosome binding site combinations. This posed two problems, first that determining which promoter:RBS combination to use would require extensive modeling, and second that creating a plasmid or BAC containing all 38 TX-TL components may not be feasible. A recent publication has shown that expressing all 38 components from a BAC was not feasible, and even expression of the components of three separate plasmids proved difficult [2].
Even before the previously mentioned paper was published, we had settled on having only 5-6 components encoded per plasmid, resulting in a 7-8 plasmid system. The TX-TL components on each plasmid would be based on relative final concentration required and functional role, in addition to being under the control of specific promoter:RBS combinations such that the relative ratio of each protein expressed on that plasmid would be correct. Each plasmid would be individually transformed into E. coli BL21 DE3 and expressed. This allows for more cells expressing the high concentration TX-TL components to be mixed with lower amounts of cells expressing the components needed at a lower concentration. The resulting purified components should be at the correct relative concentration if the correct amount of cells with each plasmid are mixed.

tRNA
In nature, the 20 standard amino acids are encoded in the DNA by 1 - 6 unique codon sequences, each of which has a corresponding tRNA molecule. In our system, we aim to have one corresponding tRNA for each amino acid to reduce complexity and ensure each amino acid can be incorporated.
To test the RNA purification method we have created a construct encoding for tRNAPhe. An ideal tRNA to use as it has been extensively studied compared to other tRNA molecules.

Once cell lysate containing overexpressed tRNA and MS2BP is added to a Ni column, the MS2 hairpins will bind to the MS2BP, which will then bind to nickel with high affinity. A DNA oligo complementary to the spacer and scar will be added and will anneal to this region, resulting in a short DNA-RNA hybrid region. Following oligo binding, RNase H will be added. RNase H will selectively cleave the RNA in this DNA-RNA hybrid. Subsequently, tRNAPhe with the correct 3’ end will be released into solution allowing for the isolation of tRNA from the MS2 hairpins and MS2BP by separating the supernatant from the nickel column.

Ribosomes
The central component of translation is the ribosome. The ribosome is a large complex comprised of three ribosomal RNAs (rRNAs) and over 50 proteins in E. coli. As such, it posed a challenge in designing a simple purification strategy.
With the TX-TL components we could tag each protein with a hexahistidine tag to purify each individually. With the ribosome, tagging each ribosomal protein and the rRNA does not ensure that functional ribosomes will form after the purification. To reduce the purification of nonfunctional ribosome components we decided to utilize a previously published ribosome purification strategy that incorporated the MS2 hairpin into the rRNA genes [3].
One MS2 hairpin is inserted into both the 23S and 16S rRNA gene at regions that are surface exposed and nonfunctional in the 3D structure. This allows for a histidine-tagged MS2BP to bind the MS2 hairpin of a fully assembled ribosome to all for a one-step purification using nickel sepharose affinity chromatography. To ensure we purify both subunits of the ribosome the MS2 hairpin will be added into the gene of both subunits rRNA gene.

Validation Construct
To validate the functionality of our TX-TL system and to provide a measure for Next vivo quality control, an additional construct was designed.

The construct consists of a T7 promoter (BBa_I719005), a medium RBS (BBa_B0034), the coding sequence for enhanced yellow fluorescent protein (EYFP; BBa_E0030) followed by a 3X flag tag (BBa_T2004), coding sequence for the spinach aptamer [4] and a double terminator (BBa_B0015). The entire EYFP coding sequence was codon optimized for efficient expression in E. coli.
To validate successful transcription, green fluorescence is observed following addition of the fluorophore 3,5-difluoro-4-hydroxybenzylidene imidazolinone (DFHBI) which binds to the spinach aptamer mRNA.

To validate successful translation, yellow fluorescence is observed following expression of EYFP. Additionally, the 3x FLAG-tag at the C-terminus of EYFP provides another method of protein quantification via western blot and as an internal size marker control for the expression of any other FLAG-tagged protein.
Biocontainment
While Next vivo is currently designed for in lab use, we also designs to make it amenable for use outside of the lab where biocontainment becomes a much larger issue. To avoid accidental contamination of the environment, we sought to have Next vivo utilize an orthogonal codon table requiring input sequences specific to this system. This would ensure that if the DNA or RNA input message were to end up in the environment, no organism would be able to properly translate the message into a functional protein. Likewise, any environmental DNA contamination in the Next vivo system would result in a non-functional protein if translated at all.

To implement such a feature we aim to change the anticodon sequence of each tRNA encoded in our system to an alternate codon. Thus, upon purification we are left with just an orthogonal anticodon tRNA pool ensuring that the purified Next vivo system requires an orthogonal codon input sequence. Additionally, with all the tRNA encoded on one plasmid, the creation of multiple orthogonal tRNA pools could easily be created. Creating orthogonal codon tables can provide a unique form of biocontainment not easily achievable with living organisms, yet raises some interesting biosafety concerns which we discuss here.


Coming soon




Each plasmid would contain 5-6 genes that are required at similar final concentrations. This enables users to express considerably less cell strains for the production of the Next vivo TX-TL components saving time and resources

Current tRNA purification design requires additional steps following the Ni-Sepharose purification. Using a MS2BP-RNase H fusion protein that positions the RNase H near the desired cut-site allows for the tRNA to be eluted by the addition of the DNA oligo making it a one step elution as well.

The base Next vivo system provides all the components for basic transcription and translation, but it might not be ideal for those tricky protein or RNA products you want to make. Several additional factor sets can be added to the Next vivo system to facilitate difficult translation processes including:
- the heat shock factor package that aid in the proper folding of large or unstable protein products; or
- the orthogonal tRNA aminoacylation package that allows specific tRNA to be labeled with non-standard amino acids which are then incorporated into your protein products providing new properties to biomolecules!

The TX-TL components in the Next vivo system will be replaced with those from two thermophilic organisms allowing TX-TL at higher and lower temperatures with the same level of protein or RNA output!


These Next vivo design features are not currently available but are some of the wild ideas and improvements that we foresee for the Next vivo TX-TL system!
References
- [1] Wang, H.H., et al., Multiplexed in vivo His-tagging of enzyme pathways for in vitro single-pot multi-enzyme catalysis. ACS synthetic biology, 2012. 1(2): p. 43-52.
- [2] Shepherd, T.R., et al., De novo design and synthesis of a 30-cistron translation-factor module. Nucleic Acids Research, 2017. 45(18): p. 10895-10905.
- [3] Youngman, E.M. and R. Green, Affinity purification of in vivo-assembled ribosomes for in vitro biochemical analysis. Methods, 2005. 36(3): p. 305-12.
- [4] Strack, R.L., M.D. Disney, and S.R. Jaffrey, A superfolding Spinach2 reveals the dynamic nature of trinucleotide repeat-containing RNA. Nat Meth, 2013. 10(12): p. 1219-1224.