Team:AQA Unesp/Applied Design
iGEM AQA_Unesp
design
applied design
Insubiota would change this method completely: the final product is a probiotic, in other words, the microrganism itself. That means that there's no need for expensive purification steps, you just need to grow the microrganism and lyophilize it to be used.
Second, Insubiota changes completely the insulin therapy: the person would not need to take several shots of insulin. Once lyophilized, the product can be mixed to a beverage or a food and be consumed. Once in the gut, the probiotic will start producing the required insulin according to glucose levels. This would be very useful and would improve the quality of life of many people that suffer with diabetes, specially children.
We have applied different concepts of synthetic biology, genetic engineering, molecular biology and microbiology to design a complete solution. Insubiota includes a glucose-responsive regulatory system based on small RNA; an insulin analog capable of being produced by bacteria and be absorbed by the intestinal cells; and a kill switch system based on the CRISPR-Cas9 regulated by light, all this in a probiotic chassis that was chosen to be the most suitable for insulin delivery.
Our name, Insubiota, comes from the words insulin and microbiota the two basis of our project and the two things that we wish to cause a complete change: the insulin therapy and production and the way we see microbiota.
chassis
As we were using Lactococcus for the first time, as a plan B, we decided to use a well established microorganism on our lab: Bacillus subtilis, that is also considered a probiotic [3]. We chose K07 strain, that is known for not producing extracellular proteases, which is indispensable for the project idea. Two vectors were used for this microorganism: pBS1C (BBa_J179000) for homologous integration and pLIKE-rep as a replicative plasmid [4].
References
[1] MAXMEN, Amy. Living therapeutics: scientists genetically modify bacteria to deliver drugs. 2017.
[2] PINTO, Joao PC et al. pSEUDO, a genetic integration standard for Lactococcus lactis. Applied and environmental microbiology, v. 77, n. 18, p. 6687-6690, 2011.
[3] DUC, Le H. et al. Characterization of Bacillus probiotics available for human use. Applied and environmental microbiology, v. 70, n. 4, p. 2161-2171, 2004.
[4] TOYMENTSEVA, Anna A. et al. The LIKE system, a novel protein expression toolbox for Bacillus subtilis based on the liaI promoter. Microbial cell factories, v. 11, n. 1, p. 143, 2012.
▲ top
control
Carbon catabolite repression (CCR) is a regulatory mechanism by which bacteria regulate the expression of functions for the use of secondary carbon sources in the presence of a preferred carbon source. Whereas the transcription factor cyclic AMP receptor protein (CRP) is responsible for CCR in gram-negative bacteria, in gram-positive bacteria the CCR is mediated by the carbon catabolite control protein A (CcpA). In L. lactis, a transcriptomic analysis revealed that Ccpa regulates genes of carbon metabolism and also its own expression [1].
To cause CCR, CcpA must bind to a specific palindromic sequence in the promoters regions of catabolic operons, called catabolite responsive elements (cre sites) [2]. Figure 1 shows the general CCR in gram-positive bacteria.

Figure 1. Phosphorylation of HPr occurs when intracellular concentrations of fructose-1,6-bisphosphate (FBP) and ATP are high, which reflects the presence of preferred carbon sources. The phosphorylated HPr(His-P) binds to the CcpA protein and this complex binds to cre site on the DNA and then represses the transcription of catabolic genes [2].
The CCR mechanism in L. lactis works as an inverter (NOT gate): when there is glucose, the gene expression is repressed, and when there is no glucose, the gene expression is activated. In order to regulate the expression of insulin in our genetically engineered machine by glucose we needed to invert this system, so we designed a circuit using the CCR mechanism regulating the expression of a small RNA (sRNA) that blocks the translation of insulin. That way, when there is glucose, the sRNA will not be expressed and the insulin will be expressed. When there is no glucose, the sRNA will be expressed and it will repress the expression of insulin. Figure 2 shows the logic of our circuit.

Figure 2. (A) The truth table of our circuit. (B) Our circuit works as a double inverter: the first inverter gate is the CCR mechanism and the second is the sRNA regulation. The input is glucose and the output is the insulin.
In order to be able to test and detect the signak from our sytem, we used the sfGFP(Bs) (BBa_K2270010) as gene reporter, previously inserted into the pSEUDO plasmid [4], which was used in our constructions. So, we designed the sRNA to target the sequence of the sfGFP(Bs) mRNA from the RBS sequence to the start codon to ensure its specificity to the target and an efficient hybridization.
To design the sRNA, we used the software RiboMaker, which gave us the sequence, hybridization energy values and the possible secondary structure of the sRNA. Analyzing the data, we selected the best two results and then performed a new analysis using the software NUPACK. By this new analysis we were able to choose the best sRNA and build our final construction. To learn more, see our modeling section.
Our final construction (BBa_K2270008) was then composed by:
- the gal operon promoter from L. lactis, which contains the cre site and has already been well studied [3];
- the designed sRNA;
- the terminator from Escherichia coli rrnb and Bacillus subtilis rrnb;

Figure 3. Our glucose-responsive control system. (A) When glucose is present the carbon catabolite repression system will repress the sRNA expression, leaving the GFP mRNA free to be translated into protein. (B) When glucose is absent, there will be no carbon catabolite repression and the sRNA will be expressed. The sRNA then will hybridizes to the GFP mRNA and block its translation.
References
[1] Zomer et al. Time-Resolved Determination of the CcpA Regulon of Lactococcus lactis subsp. cremoris MG1363. Journal of Bacteriology. vol. 189, p. 1366-1381. American Society for Microbiology, 2007.
[2] Görke, B.; Stülke, J. Carbon catabolite repression in bacteria: many ways to make the mos out of nutrients. Nature Reviews: Microbiology. vol. 6, p. 613-624. Macmillan Publishers Ltd, 2008.
[3] Luesink et al. Transcriptional activation of the glycolytic las operon and catabolite repression of the gal operon in Lactococcus lactis are mediated by the catabolite control protein CcpA. Molecular Microbiology. vol. 30, n. 4, p. 789-798. Blackwell Science Ltd, 1998.
[4] Pinto el al. pSEUDO, a genetic integration standard of Lacotococcus lactis. Applied and Environmental Microbiology. vol. 77, n. 18, p. 6687-6690, 2011.
▲ top
production
The therapeutic actuation of our system is determined by two steps: secretion and absorption. The secretion system is executed by secretory signal peptides, small amino acid sequences that, by targeting the translocation machinery, are responsible for protein exportation out of the cytoplasmic membrane (figure 1) [2]. We used the signal sequence usp45 for L. lactis and YncM for Bacillus subtilis (figure 3).

Figure 1. Signal peptide mechanism of action. When associated with the SCI-57, the signal peptide will interact with the translocation machinery on the plasma membrane and the insulin will be secreted into the extracellular environment.
After the secretion to the extracellular site, the SCI-57 must be absorbed by the enterocytes, passing through its membrane and finally reaching the bloodstream. The absorption step is accomplished by the association of the SCI-57 sequence with Cell-Penetrating Peptides (CPPs), also known as Protein Transduction Domains (PTDs). The CPPs are able to carry peptides and proteins inside the cells by passing through its membranes and are classified as cationic peptides, hydrophobic peptides and cell-type specific peptides (figure 2). We chose two CPPs for the expression design: penetratin, that demonstrated a 35% enhancement at intestinal absorption when co administered with insulin, according to Kamei et al. (2013) [3] and PenShuff, a synthetic CPP designed by Kristensen et al., 2015 [4]. In order to evaluate the influence of CPP-insulin associated position at the protein absorption, we designed N-terminal and C-terminal CPP-insulin combinations, as shown at image 3. Furthermore, we added His-tag gene sequence at one construct for further purifications.
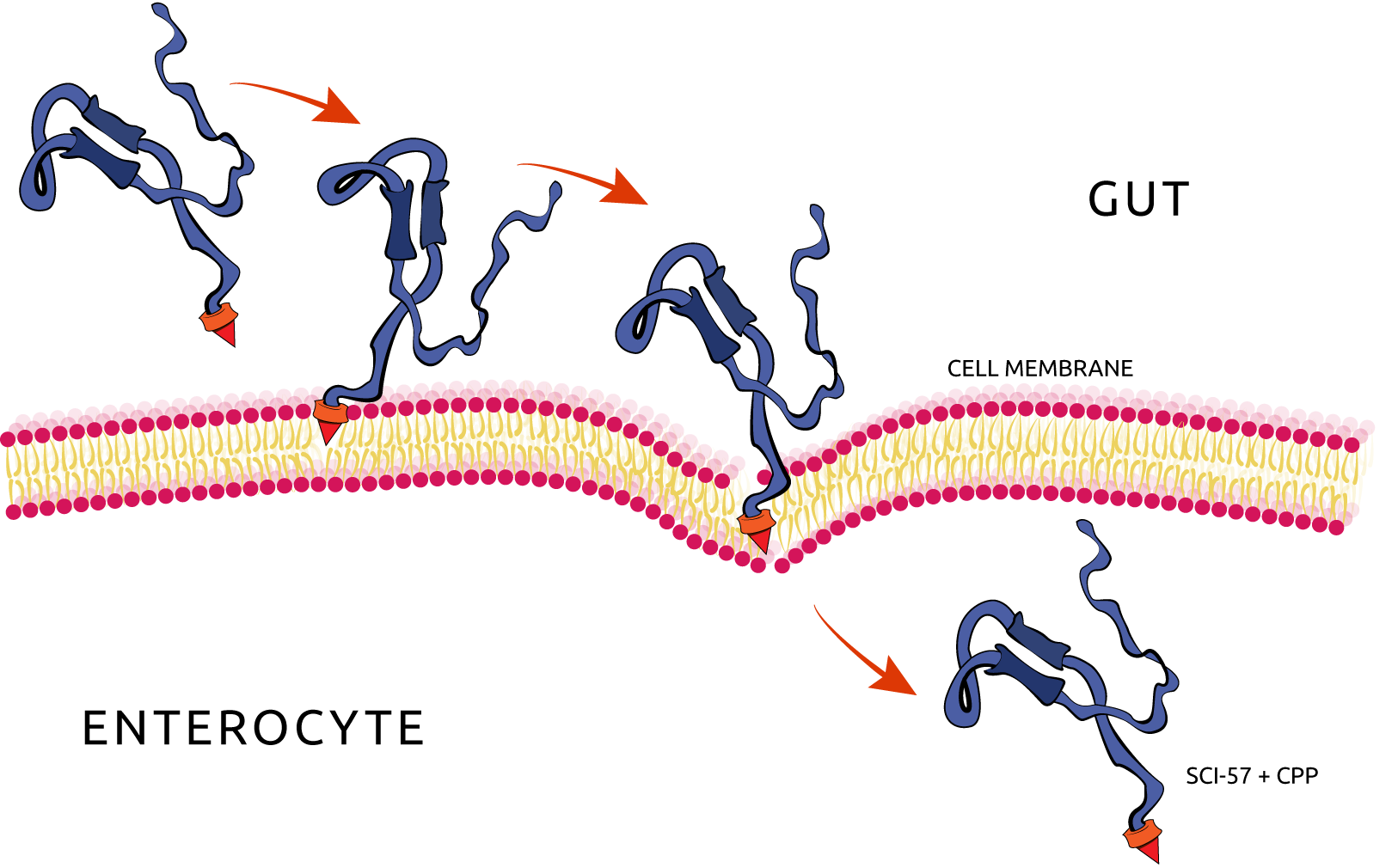
Figure 2. Insulin penetrating the enterocyte plasma membrane, by the action of the CPP/PTD.
Summarizing the expression scheme, the single chain insulin analog will be expressed by the bacteria (Lactococcus lactis and Bacillus subtilis) adhered at the gut mucus and exported outside of the cell by the action of signal peptides, reaching the gut. After that, the expressed insulin will penetrate the enterocytes’ plasma membrane by the help of associated CPPs to finally reach the bloodstream.
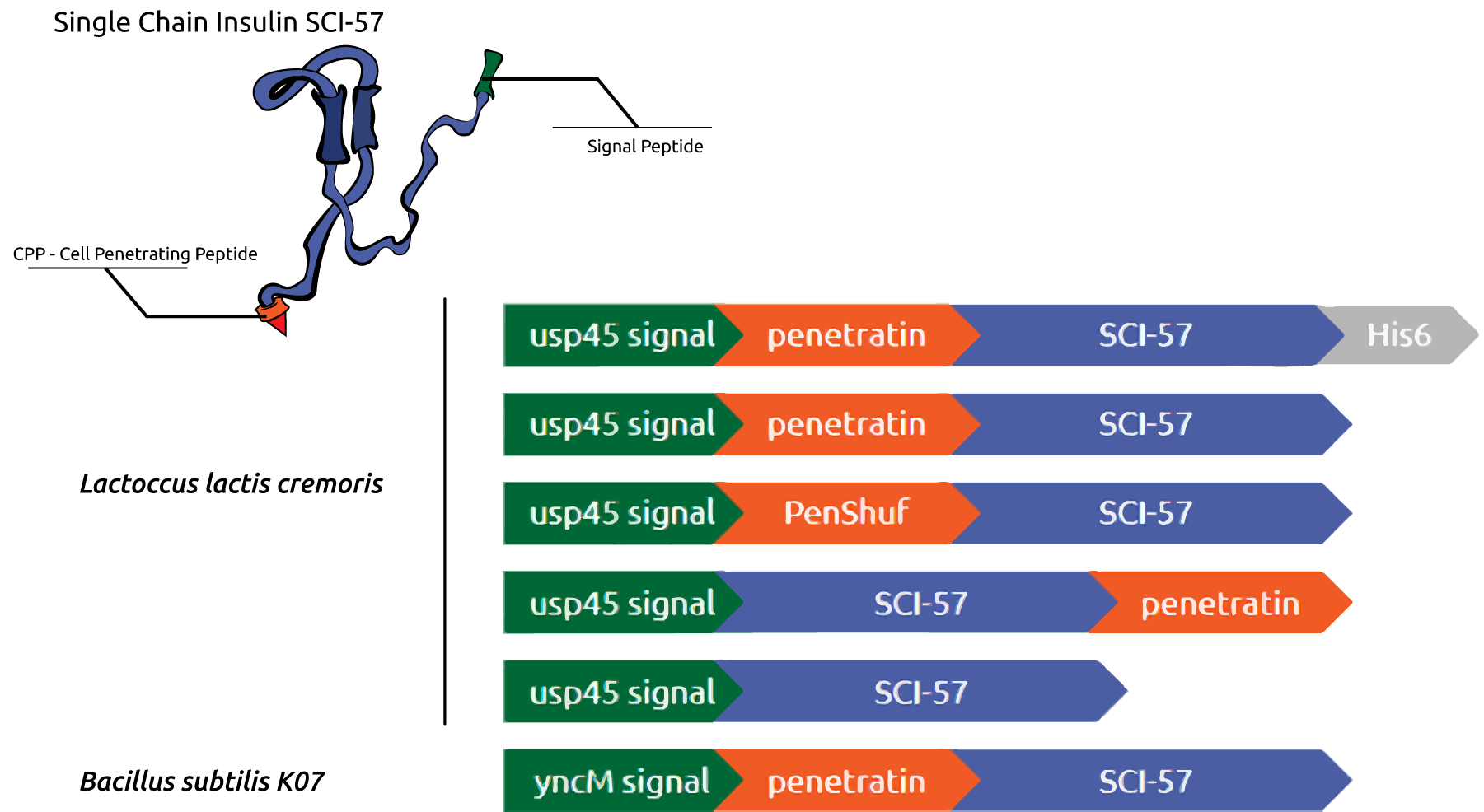
Figure 3. Insulin-CPP association gene design for Lactococcus lactis and Bacillus subtilis. Promoter and signal peptide sequence in green, CPP sequence in orange and Single Chain Insulin Analog (SCI-57) gene sequence in blue.
References
[1] HUA, Qing-xin et al. Design of an Active Ultrastable Single-chain Insulin Analog SYNTHESIS, STRUCTURE, AND THERAPEUTIC IMPLICATIONS. Journal of Biological Chemistry, v. 283, n. 21, p. 14703-14716, 2008.
[2] PETERSEN, Thomas Nordahl et al. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nature methods, v. 8, n. 10, p. 785-786, 2011.
[3] KAMEI, Noriyasu et al. Noninvasive insulin delivery: the great potential of cell-penetrating peptides. Therapeutic delivery, v. 4, n. 3, p. 315-326, 2013.
[4] KRISTENSEN, Mie et al. Penetratin-mediated transepithelial insulin permeation: Importance of cationic residues and pH for complexation and permeation. The AAPS journal, v. 17, n. 5, p. 1200-1209, 2015.
▲ top
kill switch
Our system was designed to achieve high specificity, low noise and being lethal to the cell when activated. Because our expression system is designed to function within the microbiota, it will not have light exposure, thus, prolonged exposure to light can be used as a sign that the microorganism is growing in another environment.
The composition of our kill system is divided into two parts. The fusion of CRY2 and CIB1 light-induced heterodimerizing proteins with split Cas9 (C-terminal and N-terminal) domains.
The main purpose of the construction of the kill switch was to prevent the release of our device in nature, to do that, we choose a light-inducible protein interaction modules based on Arabidopsis thaliana cryptochrome 2 and CIB1. These two proteins are able to dimerize on blue light exposure [1].
This system has been used to control gene expression as the light-activated CRISPR-Cas9 effector (LACE). Where through the binding of the units of this proteins to transcription factors and to a programmable enzyme like dcas9 represented into the scheme in figure 1 from NEU-China team. This system is capable of creating a guide and programmable system to find a specific sequence of DNA and control your gene expression [2].
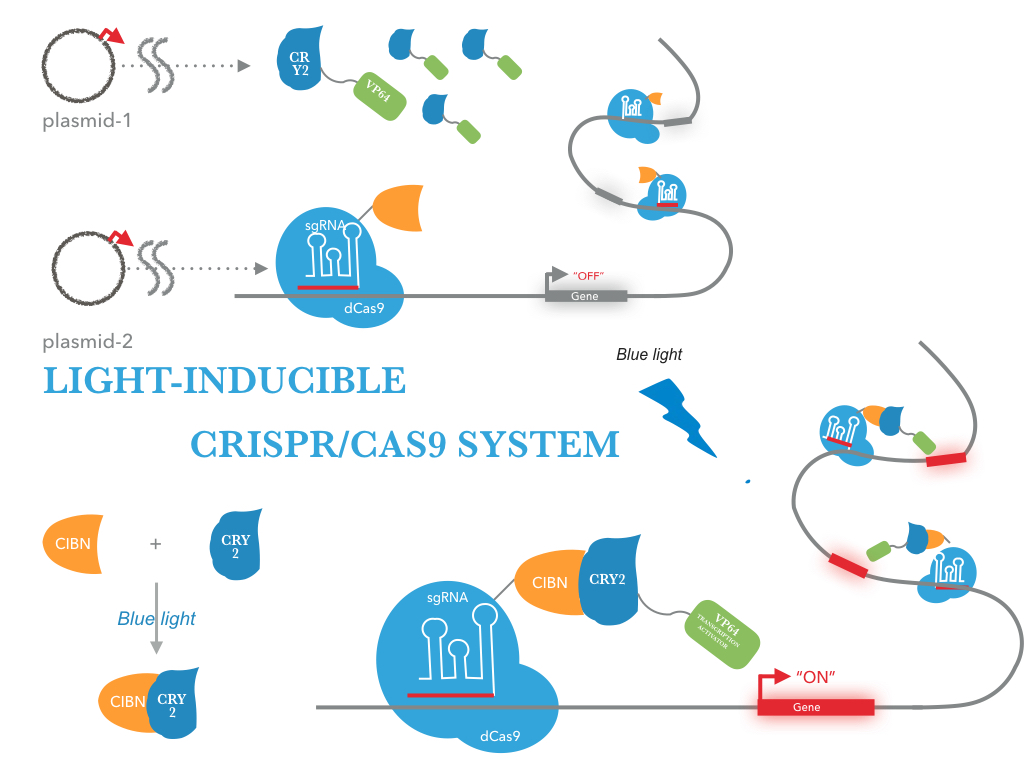
Figure 1. Light-activated CRISPR-Cas9 effector (LACE) proposed by the NEU-CHINA team 2016. At the top is the representation of the expression system; Below, on the right is the configuration used to join dCas9 to the light-induced proteins, CRY2 (BBa_K1592015) and CIB1 (BBa_K1592016) . On the left, the representation of the junction of this activated proteins in presence of the light [2].
Based on this idea we proposed a system that uses split cas9 to make a programmable and guide system to gene knock out and so kill the cell in presence of light. The CRISPR-associated protein 9 (Cas9) is a protein from Streptococcus pyogenes used in genome editing. This protein is able to target specific targets being guided by a small sequence of RNA (gRNA). To find your target this enzyme needs protospacer adjacent motif (PAM) positioned downstream of the DNA recognition site [3].
Based on the studies of Dacheng et al., we were able to propose the construction of our programmable and guide system activated by light named LAsCE (Light-activated SplitCas9 Efector) represented in figure 2.

Figure 2. Representation of the necessary constructions for the light-activated split system Cas9 Efector. Ligation of the C-terminal domain of cas 9 with the CRY2 protein and the N-terminal domain of the CIB1 protein. When the light stimulates the cells, the proteins CRY2 and CIB1 will join allowing the junction of the domains of split Cas9.
We used two software to find targets and design the gRNAs. We were capable to create a list of possibilities for the kill switch. Using the KEGG (Kyoto Encyclopedia of Genes and Genomes) and CHOPCHOP (CRISPR/Cas9 and TALEN web tool for genome editing), we model the sequences in figure 3 . The sequences were chosen to B. subtilis due to the issues we had to work with L. lactis. We decided to build and test the system first in an easier chassis to handle with, and then move it to L. lactis.

Figure 3. Proposed targets based on the metabolic pathways of Bacillus subitilis available from the KEGG database: dnaN (related to pyrimidine metabolism), rpoC (RNA polymerase unit), DNA ligase (function in DNA repair). Sequences estimated by CHOPCHOP software.
References
[1] Kennedy, Matthew J. et al. Rapid Blue Light Induction of Protein Interactions in Living Cells. Nature methods. vol.7, p.973–975. 2010.
[2] Light-inducible CRISPR/Cas9 system. NEU-China: available in << https://2016.igem.org/Team:NEU-China>>
[3] Ma, Dacheng, Shuguang Peng, and Zhen Xie. Integration and Exchange of Split dCas9 Domains for Transcriptional Controls in Mammalian Cells. Nature Communications. vol 7. 2016
▲ top
SPONSORS

WHERE TO FIND US
São Paulo State University (UNESP)
School of Pharmaceutical Sciences
Araraquara, São Paulo, Brazil
![]()
![]()
![]()