Team:Nanjing NFLS/Project

Fortified Aniline Killer.
Aniline compounds generate severe pollution problem in water and soil environments. Aniline dioxygenase is a crucial enzyme in aniline degradation process. Currently screened bacteria strains that could degrade aniline don’t take up a wide degradation spectrum, and their gene expression level could be easily affected by the aniline concentration in the environment.
Based on the facts, we performed information analysis on the basic structure of aniline dioxygenase and its gene cluster. We discovered a new gene from sludge samples, named Q gene, that demonstrated a wider substrate degradation spectrum compared to wildtype YBL2 strain. We also altered the upstream promoter of Q gene and achieved constitutive expression of Q gene and subsequent gene cluster, therefore improving the level of degradation activity of aniline dioxygenase. Through our work, we aim to lay a foundation for the bioremediation of environmental pollution.
Aniline dioxygenase gene cluster and Q gene promotor mutation
Aniline is a type of toxic synthesized organic chemical raw material, and aniline waste water treatment methods have received considerable attention. Particularly, the biological treatment utilizes microbial metabolism to degrade aniline into harmless substances (current screened bacteria strains belong to Pseudomonas, Rhodococcus, Norcadia, Acinetobacter, Alkaligenes, etc.)
The microbial degradation of aniline mainly contains aerobic and anaerobic processes. The former utilizes oxygen and oxidizes aniline to catechol under the action of aniline dioxygenase, and then releases NH4+. Catechol then serves as substrate and undergoes metabolism through meta and potential metabolic pathways. After a series of reactions, pyruvic acid and aldehyde are produced and enter the Kreb’s cycle. Basic degradation pathways are shown as follows. Figure 1 shows the degradation of aniline compounds into catechol, and Figure 2 shows the final catabolism of catechol.

Fig 1: Aniline degradation pathway
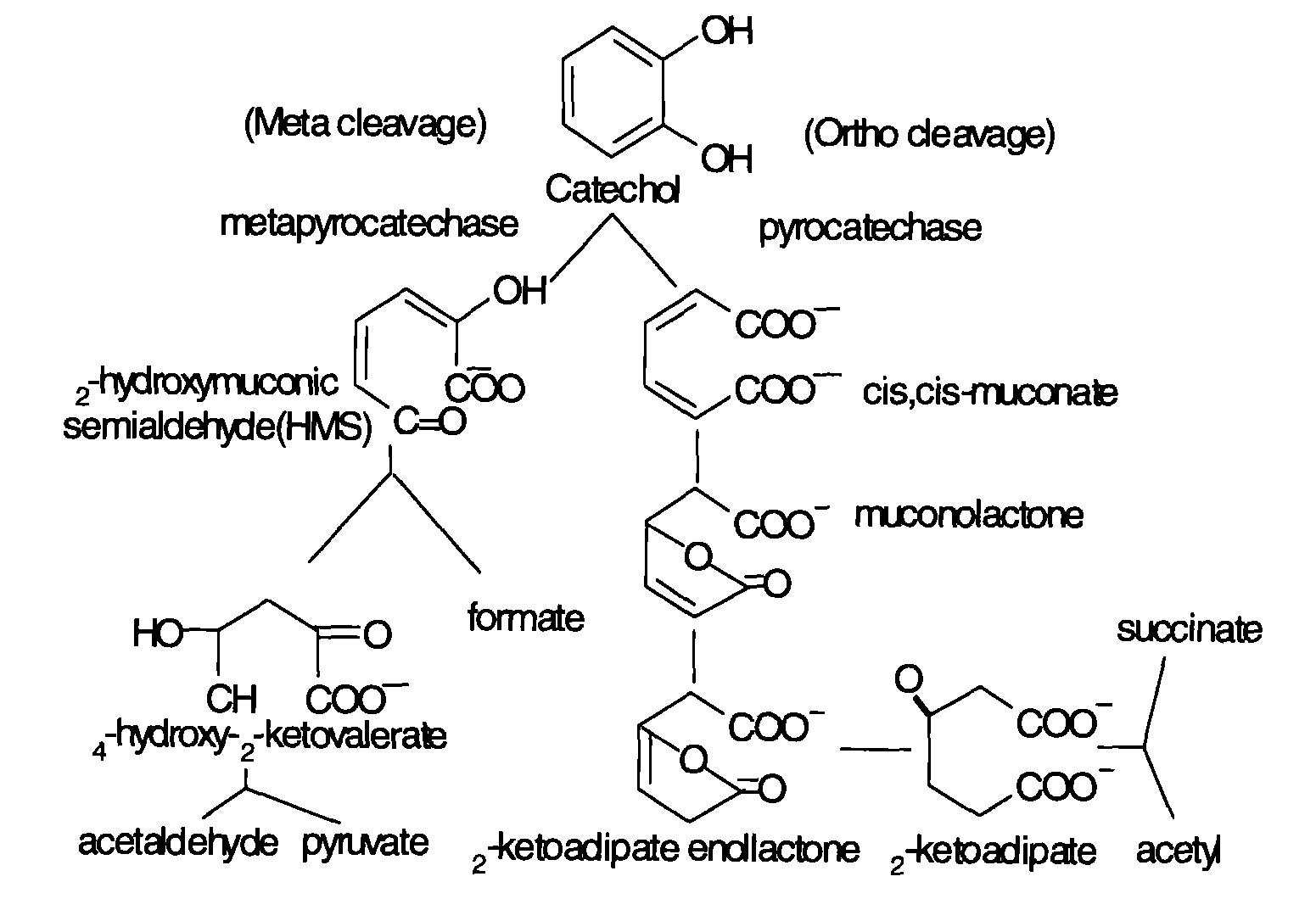
Fig 2: Aniline degradation pathway
Research has shown that bacteria genes related to aniline degradation are closely connected and arranged in clusters. Such gene clusters can turn aniline completely into intermediate metabolites of Kreb’s cycle. As mentioned before, aniline dioxygenase changing aniline into catechol is the first and foremost step of the whole reaction. Gene cluster tdn was discovered in aniline degradation plasmid pTDN1 of Pseudomonas putida UCC22. (Similar gene structures can be found in the tdn gene cluster of Acinetobacter sp. YAA and the tad gene cluster of Delftia tsuruhatensis AD9.) Genes Q and T are related to the transfer and release of amino-group; genes A1 and A2 code for the large and small subunits of aniline dioxygenase; gene B code for reductase; R is a regulatory gene (Type LysR).

Fig 3: Aniline dioxygenase structure
TadR protein is a LysR type regulatory protein. It contains two binding domains: the N terminal is DNA binding domain, and the C terminal can be combined with inducers. The Q gene promoter is induced by aniline and has positive regulation pathways. The other genes’ functions are not determined yet.
The promoter region of Q gene could bind to regulatory protein R and thus affect the expression of aniline degradation gene cluster. Without aniline induction, regulatory protein R would bind to the promoter region of Q gene and suppress the expression level of Q gene and subsequent genes. However, when the aniline concentration is too high, the expression of the gene cluster may be suppressed as well. In order to achieve constitutive expression of higher efficiency, we aim to change part of the promoter’s sequence through random mutation to lower the binding efficiency of R protein, therefore improve the expression level of the gene cluster.
gttctgctccatcttcatcctctcctgagcttatagccctgttttcacgagaccgccctcccaacggccggtctgtattcagctttccggattgtccgccggcctctttgcacgctga
tcagcaacgaacgcatccgcgatagcccggcgcgcggccggaacctcacggatcgcgattggtgacgggtataatctggaccggcaaaaccgttccgtcaatctaaaa
acgtttttgaatacaagaaaacgactatgactttttgctgttcggcccacggatgcgctatc
Q gene promotor sequence after mutation:
gttctgctccatcttcatcctctcctgagcttatagccctgttttcacgagaccgccctcccaTcggccggtctgtattcagctttccggattgtccgTcggccggtctgtattcagct
ttccggattgtccgAcggcctctttgcacgctgatcagcaacgaacgcatccgcgatagcccggcgcgcggccggaacctcacggatcgcgattggtgacgggtataatc
tggaccggcaaaaccgttccgtcaatctaaaaacgtttttgaatacaagaaaacgactatgactttttgctgttcggcccacggatgcgctatc
Sequence comparative analysis shows that two bases changed, one from A to T, the other from C to A. (The promoter region DNA is non-coding DNA mainly used for binding regulatory protein and doesn’t express target protein, so the change in amino acids is not considered here.) Results shows that two bases changed in the range of 100bp. Since the region is key to regulating protein binding, the change would affect the binding strength of regulatory protein R and weaken the affinity between the R protein and the promoter region, therefore improve the expression level of Q gene and subsequent genes. Accordingly, we could improve the gene expression efficiency by changing the promoter sequence.
Bridge PCR of Q gene and mutated promotor
The mutated promotor is connected with the Q gene sequence. As there was overlap at the two ends of products during primer design, we used bridge PCR extension to connect two ends products. The basic mechanism is shown as follows:

Fig 4: Connection strategy of promotor and Q gene
Construction of recombinant plasmid
Here’s the plasmid profile:

Fig 5: Profile of pET30a
The two gene fragments both had a primer during PCR amplification, which pre-designed and synthesized the restriction site region. Accordingly, we performed HindIII and NdeI double enzyme digestion on the vector and PCR product, and then connected it with Q gene. The clones were transformed into E.coli DH-5α competent cells, and screened through kanamycin, resulting in a number of converters.

1.Meta-genomic DNA extraction
Take samples of activated sludge in chemical pollution environment. Extract meta-genomic DNA from sludge samples using PowerSoil DNA Isolation Kit.
2.Random mutation in promoter sequence and the bridge connection of Q gene
1) Use PCR to isolate and amplify Q gene sequence. Use mutation PCR with GeneMorph 2 Random Mutagenesis Kit to isolate and amplify the target promotor sequence from meta-genomic DNA and introduce restriction enzyme sites NdeI and HindIII. In over 200 bases, two bases changed in the promotor sequence.
2) Use bridge PCR extension to connect Q gene and promotor sequences.
3) Purify amplification products from each step with Agencourt AMPure XP beads.
3.Q gene vector construction and E. coli transformation
1) Take plasmid pET30a(+) as cloning vector, link previous PCR products with plasmid pET30a(+) after enzyme digestion of NdeI and HindIII to get constructed vector.
2) Prepare chemically competent E.coli cells and transform the recombinant plasmid containing aniline dioxygenase Q gene into E.coli BL21(DE3).
3) Use Western blot and SDS-PAGE electrophoresis to verify the protein expression level.
4.Degradation bacterial strain expression through three-parent hybridization
Culture donor strain E.coli DH-5α, helper strain E.coli HB101(pRK2013) and recipient strain P.putida KT2440. Introduce recombinant plasmid pET30a(+) containing aniline dioxygenase Q gene into expression strain P.putida KT2440 through three-parent hybridization.
5.Aniline degradation test of recombinant strain Q2440
1) Prepare bacteria suspension with recombinant strain Q2440 containing Q gene.
2) Put bacteria suspension into basic salt liquid medium, and add 15 different kinds of aniline derivatives, keeping the final concentration at 20 mg/L. Use the inoculated YBL2 as the experiment group and the non-inoculated as the control.
3) Detect substrate concentration of the suspension. Substrate characterization absorption peak is at 240nm.
6.Q gene expression level verification through qPCR
Culture and collect recombinant strain Q2440 and wildtype strain YBL2. Extract RNA from the two strains using Trizol reagent and purify them with DNase I. Perform reverse transcription using 6N primer and qPCR detection.
Degradation spectrum of aniline compounds
We performed substrate degradation experiment on the three-parent hybridized recombinant bacteria strains; the inoculated YBL2 was the experiment group and the non-inoculated was the control. Three days later, the UV detection result of the bacteria suspension shows that the characteristic absorption peak at 240nm of some aniline derivatives was considerably lowered, and the peak shape was significantly changed. It could be preliminarily concluded that such aniline derivatives were degraded producing new substances. Compared to wildtype YBL2, 9 common aniline derivatives achieved more significant degradation effects (marked by red asterisk), meaning the degradation spectrum was effectively expanded; the several aniline derivatives that wildtype YBL2 couldn’t degrade were tackled as well (marked by blue “&”).

Fig 6: Degradation experiment analysis
To further verify conversion products, we selected a representative sample of bacteria suspension with peak shape change to undergo GC-MS test after treatment. Results show that both wildtype YBL2 Q gene and Q gene with modified promoter could transform methylaniline into another substance with a molecular weight of 236. It is the same molecular weight as respective Y-glutamyl aniline derivatives that was expected to be transformed from aniline compounds by Q gene. Meanwhile, no aniline derivatives or degradation product catechol was detected.

Fig 7: MS result
Level of Q gene expression
Although aniline compound degradation experiment demonstrates that the recombinant gene and the hybridized bacteria strains could degrade aniline compounds into expected metabolites, but the exact function of Q gene needs to be further examined. So we collected the bacteria and extracted the RNA after culturing the three-parent hybridized strain, and then detected the Q gene expression in recombinant and YBL2 strains when aniline induction is present and not present, respectively. Results are shown as follows.

Fig 8: qPCR
The figure shows that wildtype strains has a low gene expression level without aniline induction, and the Q gene expression considerably increased after adding aniline. Meanwhile, the modified Q gene has a high level of expression with or without aniline induction, showing that the change in promoter sequence did have an influence in regulatory protein binding. The gene clusters in recombinant strains underwent constitutive expression, no longer sensitive to changes in aniline concentration in the environment. Instead, the bacteria constantly expressed the enzymes needed to degrade aniline compounds, and are more suitable for the bioremediation of polluted environment.
Aniline degradation verification with Vorticella
Vorticella is a kind of aquatic organism that’s very sensitive to the environment. When aniline compound level in water exceeds a certain level, locomotor activity of vorticella will decrease. In order to visually and directly examine the aniline degradation ability of our engineered recombinant bacteria, we decided to add vorticella to aniline solutions treated and untreated with engineered bacteria respectively. As the video shows, in treated solution where aniline concentration is greatly lowered, the vorticella’s locomotor activity isn’t obviously affected. Meanwhile, in untreated aniline solution, the vorticella’s locomotor activity significantly decreased within a short period of time.
Download Untreated Video here.
And Download Treated Video here.


1.Untreated 2.Treated
1.Verification and quality control of metagenome
The extraction results of meta-genomic DNA from sludge in polluted areas are presented in the figure. AGE shows that main DNA fragments are longer than 15kb and can be used in subsequent experiments.

Fig 9: meta-genomic DNA electrophoresis
2.Verification of mutated promotor sequence
The promoter region undergoes PCR and AGE. The result shows that the length of fragments is consistent with target fragments, meaning the meta-genomic DNA could be amplified and contains bacteria strains for at least one type of aniline compound.
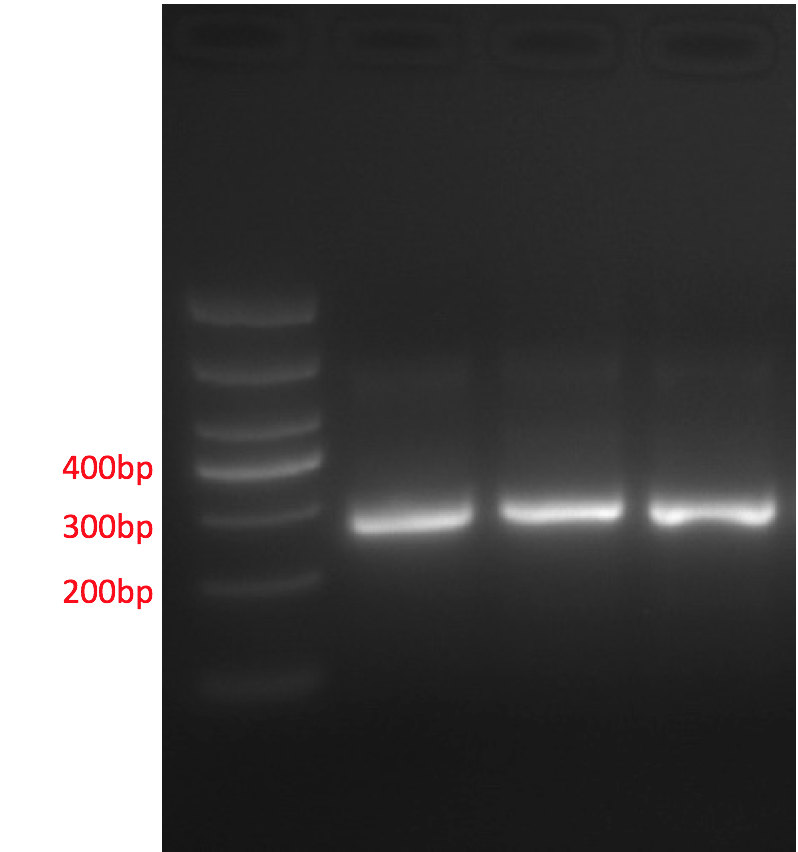
Fig 10: Promotor PCR product
In order to acquire a promoter sequence that contain mutation sites, we used the PCR amplification product of the promoter region as the template, and performed mutation amplification of medium frequency using random mutation PCR enzyme. Sanger sequencing were done on the two amplification results, and the sequence results are shown as follows.
Original Sanger sequencing:

Fig 11: Promotor Sanger diagram
Mutation site:

Fig 12: Mutation Sanger diagram
3. Verification of Q gene
The complete sequence of Q gene is also acquired through PCR amplification, electrophoresis, and Sanger sequencing. The length of fragments and the sequence both shows that the meta-genomic DNA in the sample contain aniline degradation gene sequence and could be used for further cloning and expression analysis.

Fig 13: Q gene PCR product
Sanger sequencing of Q gene:

Fig 14: Q gene Sanger sequencing diagram
4. Verification of the fusion of two sequences
We performed bridge connection between purified Q gene segment and randomly mutated promoter gene sequence, and the results are shown as follows. The target segments’ lengths are as expected.

Fig 15: Bridged part electrophoresis
5. Verification of recombinant plasmid

Fig 16: recombined plasmid
Lane 1 is the recombinant plasmid, and Lane 2 is the result of the recombinant plasmid undergoing double enzyme digestion using MluI and HindIII. The lengths of segments were the same as required, meaning the target gene has been successfully inserted into the vector, and could undergo further fusion expression and verification.
6. Western identification results of transformation products
The acquired recombinant plasmid is transferred into E.coli and cultured. The target protein was properly expressed, and its degradation function has to be further verified through three-parent hybridization. The Western blot testing results are shown as follows. Results has shown that Q gene was properly expressed in host bacteria; the protein expression contains histidine tags, and the electrophoresis results on the right were histidine antibody hybridization results, showing that the target protein was also properly expressed in E.coli.
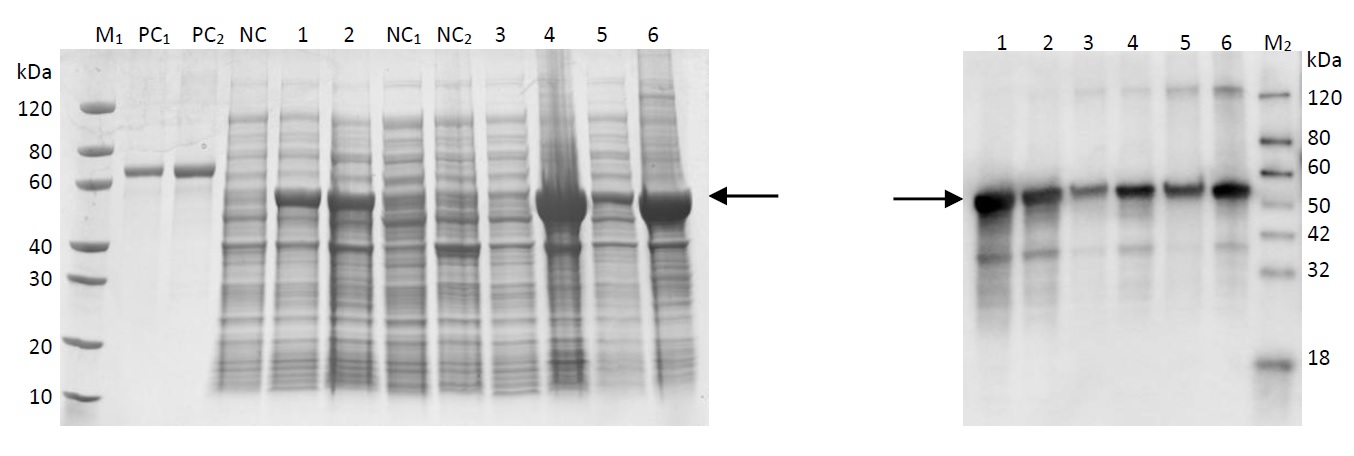
Fig 17: Western PAGE
Lane M1: Protein marker
Lane M2: Western marker
Lane PC1: BSA (1µg)
Lane PC2: BSA (2µg)
Lane NC: Undifferentiated whole cell lysate
Lane 1: Whole cell lysate, 15℃ induced for 16 hours
Lane 2: Whole cell lysate, 37℃ induced for 4 hours
Lane NC1: Supernatant of undifferentiated cell lysate
Lane NC2: Precipitation of undifferentiated cell lysate
Lane 3: Cell lysate supernatant, 15℃ induced for 16 hours
Lane 4: Cell lysate precipitation, 15℃ induced for 16 hours
Lane 5: Cell lysate supernatant, 37℃ induced for 4 hours
Lane 6: Cell lysate precipitation, 37℃ induced for 4 hours
Safety
Safety is the most crucial factor throughout all the projects. Throughout our project, we make sure our members are handling safe material in a safe environment, and following safety rules. Our PI Dr. Tu is responsible for our safety during laboratory settings.
Materials and Facilities
The laboratory we shared with graduate students of Southeast University, State Key Laboratory of Bioelectronics, is well equipped with fire hydrant, fire extinguisher and fire blanket to make sure the fire could be extinguished in urgent cases. All the team members have learnt how to use these extinguishing equipments. The laboratory has a explicit list of safety rules and regulations hung on the wall. We have all carefully read them and learnt all these instructions. In these ways, the safety of the laboratory is fully ensured.
Lab Rule
As we shared lab with Southeast University, we use the Southeast University biolab safety rules (http://sbc.seu.edu.cn/e4/89/c5586a58505/page.htm) as a reference. We also have the Chinese center for disease control and prevention (http://www.chinacdc.cn/jkzt/swaq/jdjc/) and China Biosafety information web (http://www.biosafety.com.cn/) to guide our security.
Safety Training
In order to ensure our experiments are conducted under absolutely safe condition, every member of Nanjing_NFLS has been involved in relevant training of lab safety before operating in the lab. The training includes experimental safety knowledge of molecular biology, lab techniques, introductions to regular apparatus and software, safety of standard operation, personnel protection, common emergency response and handling, etc. We have ensured that every member of our team has a full awareness of experiment safety. Only in this way can we avoid various experimental dangers by taking preventive measures in advance.
Parts
Harmful parts are not allowed and we ensure that none of our parts would raise any safety issue according to the current professional knowledge.